Pyrazole (E)-vinyl sulfone 3 was identified as a potential covalent CHIKV nsp2 protease inhibitor from a high throughput screen in the READDI Antiviral Drug Discovery Center at UNC.1 Initial re-synthesis of the hit compound yielded a mixture of acyclic (3) and cyclic sulfones (4) (Scheme 1), which were identified by 1D and 2D NMR, but could not be differentiated by LCMS and HPLC analyses.
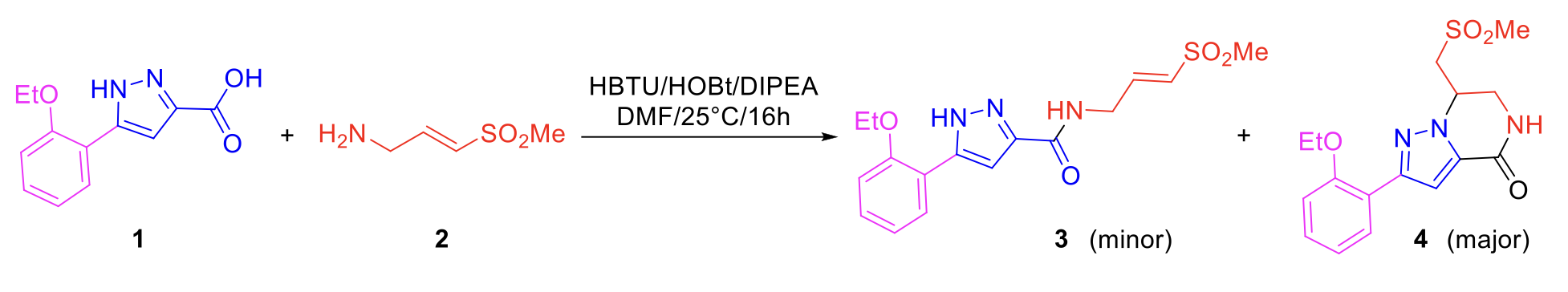
Scheme 1. Re-synthesis identifies cyclic analog 4.
The formation of the cyclic analog 4 occurred via an intramolecular Michael reaction (Scheme 2) where the acyclic analog 3 cyclizes to form the cyclic compound 4.
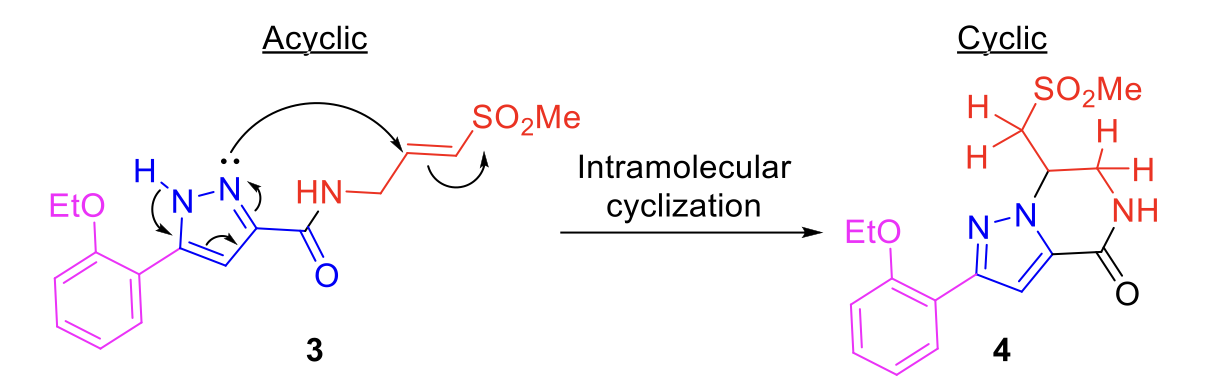
Scheme 2. Intramolecular Michael reaction favors cyclization.
NMR-based characterization of acyclic and cyclic analogs:
The structures of the acyclic (3) and cyclic analogs (4) were confirmed by 2D NMR spectroscopic data including COSY, HSQC, and HMBC spectra. 1H NMR analysis of compound 3 showed characteristic olefinic protons at δ 6.69 ppm (dt, J = 15.3, 1.8 Hz, 1H) and δ 6.82 ppm (dt, J = 15.3, 4.4 Hz, 1H) respectively (Figure 1a). The disappearance of the olefinic protons and appearance of a multiplet at δ 5.07 ppm (m, J = 8.9, 4.5 Hz, H-9) was a key point of difference for the formation of compound 4 (Figure 1b). Another key difference was the disappearance of H-12 at δ 4.11 ppm (ddd, J = 6.2, 4.2, 1.8 Hz, 2H) in the cyclic analog 4. After cyclization, H-8 appeared at δ 3.70 ppm (td, J = 6.3, 3.5 Hz, 1H) and δ 3.93 ppm (ddd, J = 13.3, 4.4, 2.3 Hz, 1H) respectively in analog 4 (Figure 1b). H-11 in analog 4 appeared as distinct protons at δ 3.76-3.72 ppm (m, 1H) and δ 3.98 ppm (dd, J = 14.4, 3.9 Hz, 1H) respectively (Figure 1b). Important to note that the H-12 protons in compound 3 appeared together at δ 4.11 ppm (ddd, J = 6.2, 4.2, 1.8 Hz, 2H) (Figure 1a). However, in the cyclic compound 4, the corresponding protons denoted as H-8 are non-equivalent (showing different signals in the 1H NMR spectra) (Figure 1b). These protons were initially equivalent in the open form (denoted by H-13 and H-14 in Figure 1a).
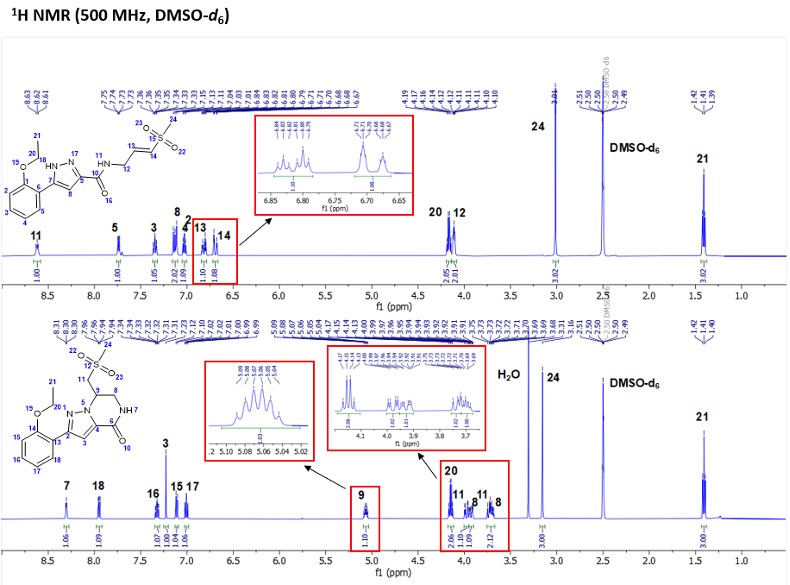
Figure 1. 1H NMR spectra of compounds 3 and 4 depicting the key distinguishable peaks between the open and cyclic analogs.
1H-13C HMBC analysis of cyclic analog 4 revealed that H-9 illustrates a three-bond correlation with atom C-4 (Figure 2). This correlation is the key evidence for formation of (E)-vinyl sulfone 4.
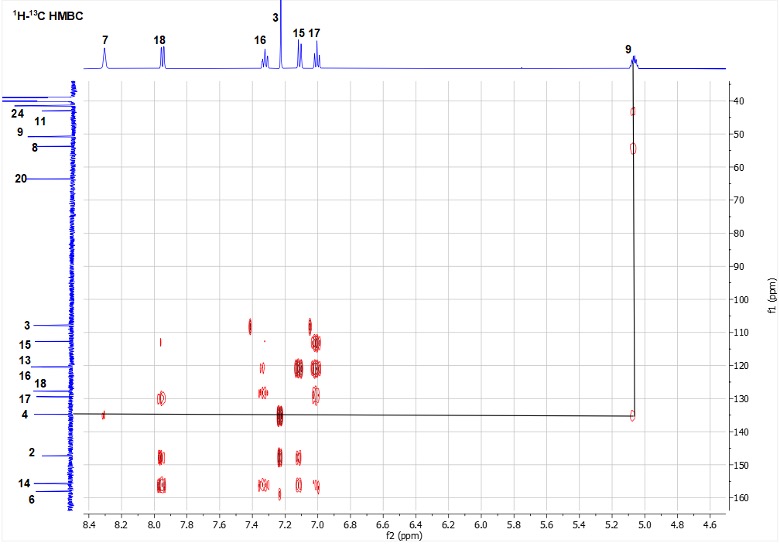
Figure 2. HMBC spectrum of 4.
Vinyl sulfones have (E)-configuration:
It is important to note that the vinyl sulfone warhead 2 was synthesized as (E)-isomer (Figure 3a), which yielded the corresponding acyclic analog 3 in (E)-configuration during the coupling reaction (Figure 3b). The (E)-isomer is confirmed in both cases from the coupling constant (J = 15 Hz) values of the olefinic protons in vinyl sulfones 2 and 3.
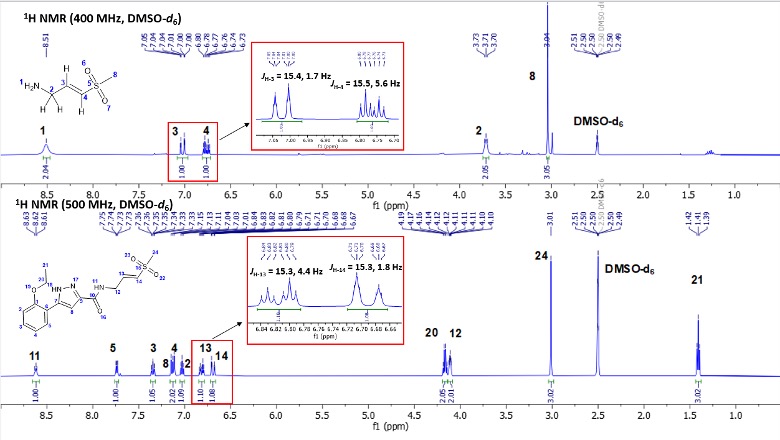
Figure 3. 1H NMR spectra of (E)-vinyl sulfones 2 and 3 depicting the olefinic protons with corresponding J values.
Although we were able to characterize the acyclic and the cyclic sulfones, full biological evaluation of the screening hit and the synthesis of analogs required the development of robust protocols to access the pure acyclic and cyclic pyrazoles.
This encouraged us to solve three major issues associated with the chemistry:
- Develop a robust analytical method to identify and separate the acyclic and cyclic analogs
- Develop a robust synthetic protocol to access the acyclic analog exclusively in high yields
- Develop a synthetic method to completely convert acyclic to cyclic analog
- Development of Analytical Method
Initial LCMS run time of 3 min and 6 min did not separate out the acyclic and cyclic analogs and they overlapped with one another in the LCMS spectra. Even on preparative (8 min) and analytical HPLC (6 min) runs, compounds 3 and 4 did not show up as distinct separable peaks. This encouraged us to develop an in-house analytical method to monitor the reactions, separate the acyclic and cyclic analogs, and identify the products. After several trials, it was observed that a run time of 12 min on LCMS (Figures 4a and 4b) and 26 min on HPLC (Figure 4c) could separate out the acyclic and cyclic analogs as two distinct peaks (LCMS column: C18 2.7 mm (Agilent); HPLC column: Luna 5 mm phenyl-hexyl (Phenomenex). Thus, the acyclic and cyclic forms were separable only with extended HPLC runs.
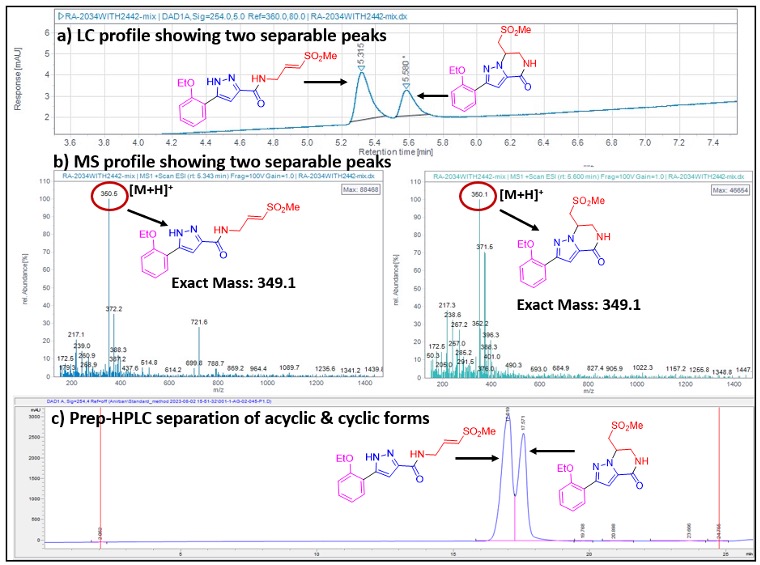
Figure 4. LCMS (4a–b) and HPLC (4c) profiles of acyclic (3) and (4) cyclic analogs.
The acyclic (3) and cyclic (4) vinyl sulfones were isolated with high purity from the HPLC purification (Figure 5).
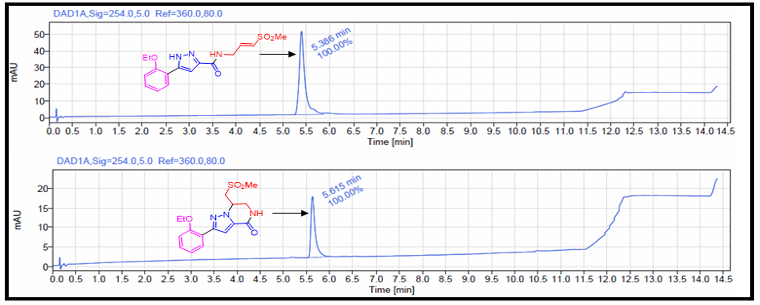
Figure 5. LCMS profiles of pure acyclic and cyclic analogs after preparative-HPLC purification.
2. Synthesis of Acyclic Analog and Methods for Cyclization
a) Optimization of Reaction Conditions for Amide Coupling:
As the amide coupling conditions were prone to form both the acyclic and cyclic analogs, it was important to reduce the propensity to form the cyclic analog. In this regard, we tried different conditions for performing the acid-amine coupling. Using pyrazole carboxylic acid 1 (1.0 equiv.), amine 2 (1.2 equiv.), HATU (1.5 equiv.), DIPEA (3.0 equiv.) in DMF at 25°C for 2h afforded the acyclic (3) and the cyclic (4) analogs in 60:40 ratio (Condition 1, Table 1). Similar ratio of formation of 3 and 4 were observed when treated with T3P (1.5 equiv.) and TEA (3.0 equiv.) in DMF at 25°C for 2h (Condition 2, Table 1). When the coupling was carried out using EDC.HCl (1.5 equiv.), HOBt (1.5 equiv.) in DMF at 25°C for 2h, ratio of acyclic (3) and cyclic (4) analogs was 70:30 (Condition 3, Table 1). Using PyBOP (1.0 equiv.), DIPEA (2.0 equiv.) in DMF at 25°C for 16h (Condition 4, Table 1) or DIC (1.2 equiv.), DMAP (2.0 equiv.) in DMF at 25°C for 16h (Condition 5, Table 1) did not improve the fate of the reaction. However, with TBTU (1.5 equiv.) and pyridine (0.2M), exclusive formation of pyrazole (E)-vinyl sulfone 3 was observed in 70% yield (Condition 6, Table 1). This optimized reaction condition will be used for coupling the substituted pyrazole carboxylic acids with the cysteine-capturing warheads during SAR exploration.
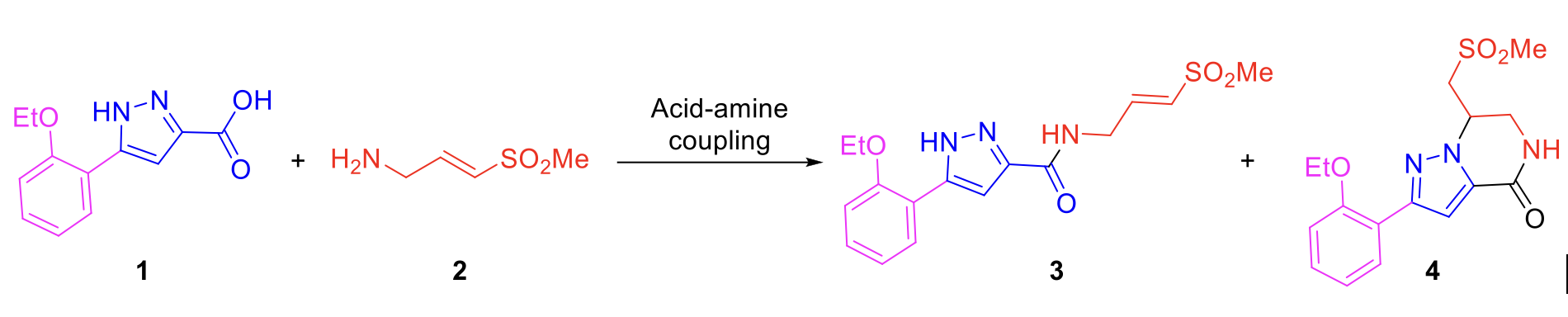
Scheme 3. Amide coupling leads to both acyclic and cyclic analogs.
Table 1. Optimization of Reaction Conditions for Amide Couplinga
| Entry | Coupling Reagent | Base | Solvent | Temp (°C) | Time (h) | 3:4b |
| 1. | HATU | DIPEA | DMF | 25 | 2 | 60:40 |
| 2. | T3P | TEA | DMF | 25 | 2 | 60:40 |
| 3. | EDC.HCl | HOBt | ACN | 25 | 2 | 70:30 |
| 4. | PyBOP | DIPEA | DMF | 25 | 16 | 60:40 |
| 5. | DIC | DMAP | DMF | 25 | 16 | 60:40 |
| 6. | TBTU | Pyridine | Pyridine | 25 | 2 | 100:0 |
aReaction conditions: 1 (1.0 eq.), 2 (1.2 eq.), solvent (0.2 M). bBased on LCMS and 1H NMR analyses.
b) Alternative Route to Access Acyclic Analogs by Protection of Pyrazole NH:
An alternate regioselective protocol using MOM-protection of pyrazole 1 was developed to enable synthesis of pyrazole analogs that were more prone to cyclization (Scheme 4). MOM (methoxymethyl) protection at the N-1 position of pyrazole 1 was achieved using 3.0 eq. of CH3OCH2Cl (MOMCl), 1.2 eq. of K2CO3 in DMSO at 25°C for 2h (Scheme 4, Step 1). TBTU-pyridine mediated amide coupling (Scheme 4, Step 2), followed by MOM deprotection yielded acyclic (E)-vinyl sulfone 3 (Scheme 4, Step 3). This strategy will be useful to access pyrazole-containing acyclic covalent inhibitors in good yields, avoiding intramolecular cyclization, and minimizing tedious purification steps. The high regioselectivity of the N-1 substituted pyrazole is illustrated by previous reports from Almena et al.2 and Huang et al3a, Norman et al3b.
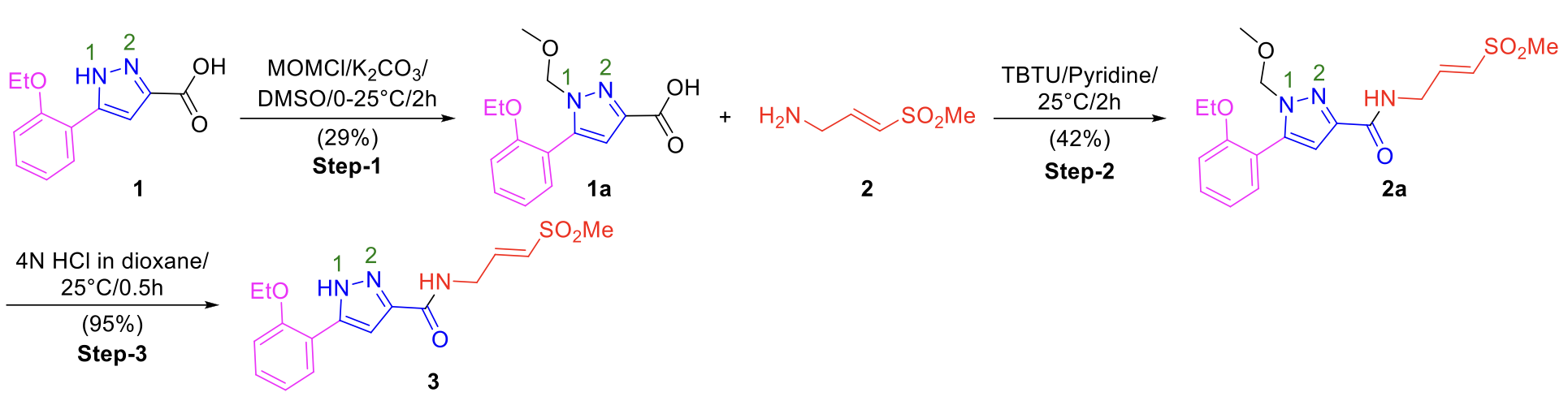
Scheme 4. K2CO3-mediated MOM-protection of pyrazole NH (Step 1), amide coupling (Step 2), and subsequent deprotection (Step 3) a,b
aReaction conditions: Step-1: 1 (1.0 eq.), MOMCl (3.0 eq.), K2CO3 (1.2 eq.), DMSO (0.3 M), 25°C, 2h. Step-2: 1a (1.0 eq.), 2 (1.2 eq.), TBTU (1.5 eq.), pyridine (0.2 M), 25°C, 2h. Step-3: 2a (1.0 eq.), 4N HCl in dioxane (10.0 eq.), 25°C, 0.5h. bYields are isolated yields.
c) Optimization of Reaction Conditions for Intramolecular Cyclization:
The optimal reaction conditions for conversion to cyclic form by intramolecular aza-Michael reaction were also identified.4 Stability studies demonstrated that acyclic pyrazole 3 is converted cyclic pyrazole 4 in mild basic media, but 4 is stable under standard laboratory conditions. Intramolecular aza-Michael reaction was performed under basic conditions with polar protic and aprotic solvents. Different bases viz. K2CO3, NaHCO3, TEA, DIPEA, DBU were used at room temperature to favor the cyclization (Table 2). Interestingly, two reaction conditions: K2CO3 (3.5 equiv.), EtOH, 25°C, 12h (Condition 1) and Na2CO3 (3.0 equiv.), dioxane:H2O (1:1), 25°C, 12h (Condition 5) afforded the cyclic analog 4 with 100% conversion. Important to mention that other bases like NaHCO3 (53% in H2O and 88% in MeOH), TEA (49% in MeOH), DIPEA (96% in DMF), DBU (92% in ACN) also favored the cyclization with good conversion rates (Table 2). However, due to the difficulty in separation of the acyclic and cyclic analogs, we favored the conditions with 100% conversion. This will avoid the need to perform difficult HPLC purifications of all the analogs for SAR studies.
Table 2. Optimization of Reaction Conditions for Intramolecular Cyclization
| Sl. No. | Base | Solvent | Temp (°C) | Time (h) | % Conversiona |
| 1. | K2CO3 (3.5 eq.) | EtOH | 25 | 12 | 100 |
| 2. | NaHCO3 (3.0 eq.) | H2O | 25 | 12 | 53 |
| 3. | NaHCO3 (3.0 eq.) | MeOH | 25 | 12 | 88 |
| 4. | K2CO3 (3.5 eq.) | H2O | 25 | 12 | 0 |
| 5. | Na2CO3 (3.0 eq.) | Dioxane:H2O | 25 | 12 | 100 |
| 6. | TEA (3.0 eq.)
|
MeOH | 25 | 12 | 49 |
| 7. | DIPEA (3.0 eq.) | DMF | 25 | 12 | 96 |
| 8. | DBU (0.5 eq.) | ACN | 25 | 12 | 92 |
aBased on LCMS analyses of crude reaction mixtures.
We are trying to understand what factors (electronics, sterics, basicity) might affect the propensity for the formation of acyclic and cyclic analogs. Accordingly, suitable modifications will be made on the pyrazole ring and the warhead to understand the relative stability between the formation of acyclic and cyclic vinyl sulfones.
References:
- https://readdi-ac.org/
- Almena et al. 1998, 35, 1263-1268.
- (a) Huang et al. J Org. Chem. 2017, 82, 8864-8872. (b) Norman et al. J Org. Chem. 2022, 87, 10018-10025.
- (a) McDowell et al. J Chem. Soc. B: Phys. Org. 1967, 0, 343-348; (b) McDowell et al. J Chem. Soc. B: Phys. Org. 1967, 0, 348-350.