The Recap
It’s been one year since the last update on the α/β hydrolase domain 2 (ABHD2) as a target for non-hormonal contraception (see my previous post here). The major challenges were the production of ABHD2 and development of a reliable activity assay to screen and quantify inhibitory compounds. At that point 14 different constructs had been tested in E. coli and 17 in BEVS, both displaying only mediocre amounts of soluble protein either in the initial screen or after scale-up that were not sufficient for the use in downstream applications. In the meantime, other ABHDs (ABHD10, 11, and 14B) were purified with the aim to develop a selectivity panel as well as an activity assay using p-nitrophenyls as general hydrolase substrates.
The goal of this project is two-fold: Characterization of ABHD2 to evaluate the protein as a target for non-hormonal contraception as well as the generation of a full target-enabling package (TEP) that offers molecular tools for further investigation of the enzyme. This includes protocols for production of soluble ABHD2, assays that test folding status and activity of the purified protein, investigation of the effect of progesterone as well as the published inhibitor (CBK600192) for a molecular characterization. On this knowledge will then be expanded to develop more potent inhibitory compounds towards a chemical probe. In parallel, crystal structures of the free protein and in complex with ligands as well as the development of a specific antibody are needed for a complete TEP.
The Results
Protein production
Production of ABHD2 was approach by multiple efforts in parallel. 120 different constructs with different truncations and terminal tags were cloned and tested in different E. coli strains (BL21(DE3)R3pRARE2, T7 express cells from NEB), 20 in BEVS, and 4 in mammalian cells at SGC Toronto and SGC Karolinska. A full list of tested constructs can be found in the Zenodo entry. In parallel, LiberumBio tested the cell-free expression of one construct with an N-terminal His-tag for residues L33-E425. Ca. 300 µg of ABHD2 with other contaminating proteins were acquired with 1 mM Tween-80 and reducing agent. Genscript was contracted with testing two different secreted constructs containing residues R31 to E425 of the protein with an N-terminal signal sequence and C-terminal His-tag in their TurboCHO system, in which the produced protein is secreted. SDS-PAGE revealed high molecular-weight aggregates of expressed protein, but no secreted protein was detected in the medium. The band of the monomeric protein could be recovered under reducing conditions, which indicated misfolding under oxidative conditions.
As an alternative approach, mutational screening using the PROSS tool was used to create three constructs with 9, 18, and 33 mutations inserted, based on multiple sequence alignment, to increase solubility and stability.1 Only small amounts (less than 1 mg) of each construct could be extracted for the constructs carrying 18 and 33 mutations, that however displayed activity in a general hydrolase activity assay.
At the same time, the team at SGC Toronto found that addition of 1% Triton X-100 to the lysis buffer could substantially increase the amounts of soluble ABHD2, based on the observation from cell-free expression, where soluble ABHD2 could retained in solution by addition of a detergent. For a construct of L33-E425 carrying an N-terminal His-FLAG tag, as described in this post by Madison Edwards, several milligrams of protein were purified. This could be reproduced for other constructs with an N-terminal His-tag as well as a His-FLAG-tag after which a TEV cutting site was inserted for tag-removal. The identity of the purified protein was confirmed by mass spectrometry.
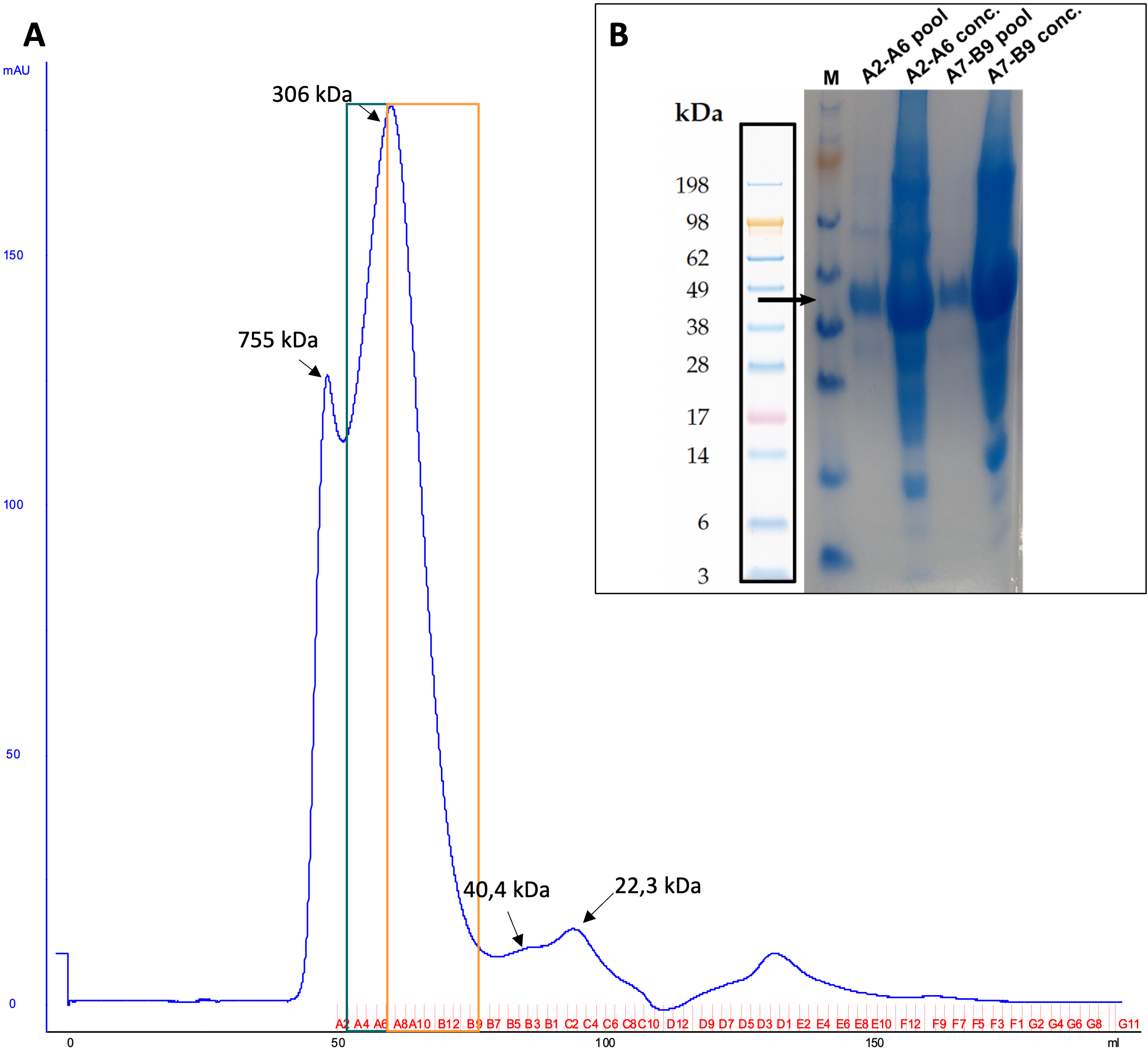
Figure 1: A) Exemplary SEC profile of ABHD2-c049 (N-His-FLAG-ABHD2(L33-E425)) on a HiLoad 16/60 Superdex 200 pg. Fractions of green and orange boxes were pooled and concentrated. B) SDS-PAGE and Coomassie stain of final samples in diluted and concentrated form are displayed, too. Theoretical molecular weight of purified construct is indicated by an black arrow.
Subcellular localization
Based on results from Genscript, which showed no secretion of the protein but only high molecular-weight aggregates under oxidative conditions, as well as the lack of disulfide bonds in the structure prediction by AlphaFold and behavior of the protein in buffer (stable in the presence of reducing agent), the extracellular localization of ABHD2 was put into question.
At SGC Frankfurt and SGC Toronto, this was investigated by transient transfection of HEK293T and U2OS cells with different full-length constructs of ABHD2, one carrying either a C-terminal MYC- or FLAG-tag, that was subsequently detected by immunofluorescence, after permeabilizing the cells. The second construct carried a C-terminal eGFP for detection of eGFP-fluorescence without immunostaining.
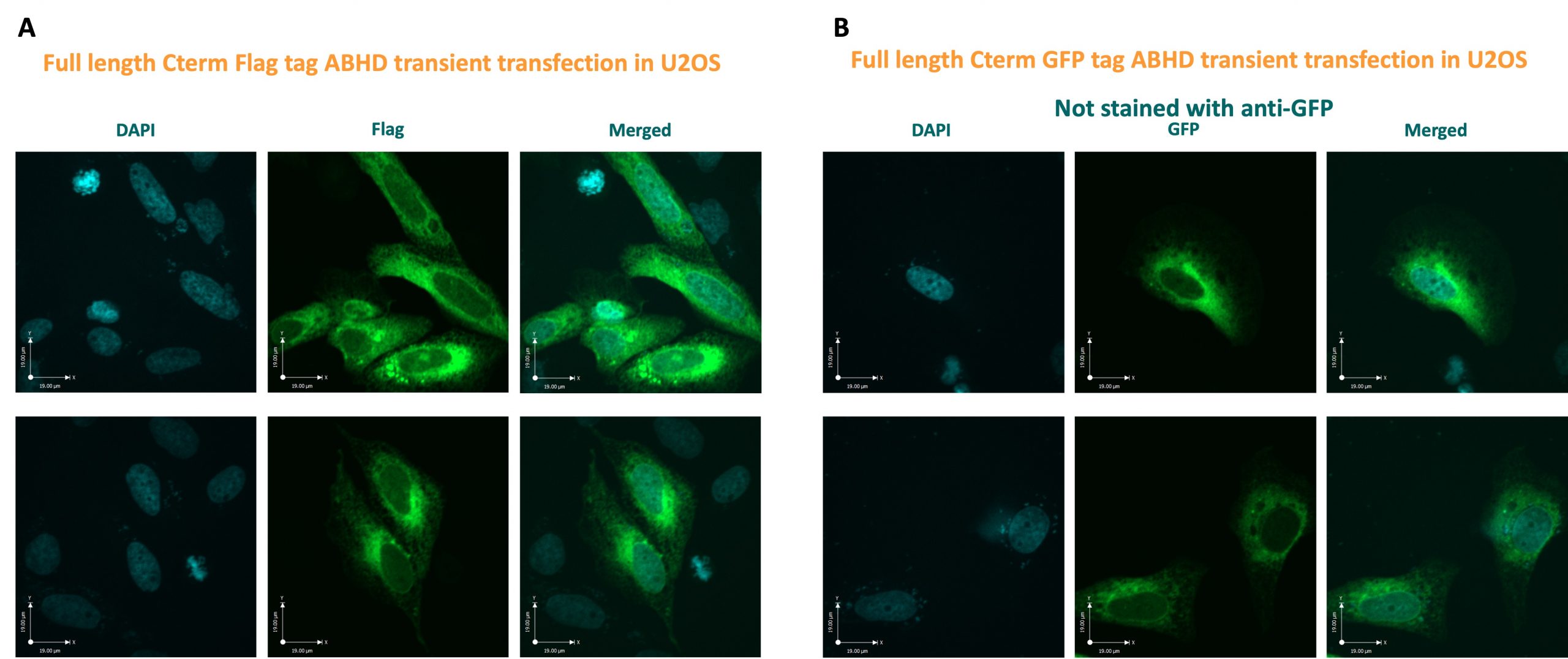
Figure 2: Confocal images of U2OS cells transiently transfected with full-length ABHD2 constructs, carrying either a C-terminal FLAG-tag (A) or C-terminal eGFP (B). A) Immunofluorescence of permeabilized cells using an anti-FLAG is shown, while in B) the intrinsic fluorescence of eGFP from untreated cells is displayed. For neither, fluorescence at the plasma membrane could be observed.
Both, immune- and eGFP-fluorescence were only detected in the cytosol, while no fluorescence was observed at the membrane, indicating no membrane-localization of ABHD2 for the transfected cells.
Folding and interactions
Folding of recombinantly purified N-FLAG-ABHD2 (L33-E425) was tested by differential scanning fluorimetry (DSF) using tryptophan fluorescence on a PrometheusTM NT.48 (NanoTemper Technologies). For this, 4 µM ABHD2 (L33-E425) were prepared in 50 mM HEPES pH 8, 500 mM NaCl, 10% glycerol, 1 mM TCEP, with and without 1% DMSO and after 5 min of centrifugation at 15,000xg transferred to capillaries PrometheusTM NT.48 Capillaries (NanoTemper Technologies). Triplicate samples were heated with 1 °C/min from 25 to 95 °C, while intrinsic fluorescence at 330 and 350 nm as well as the backscattering was recorded. A melting curve was observed from which an infliction point at 44.2 °C could be derived, corresponding to the melting temperature, indicating structural integrity of the purified protein. No significant difference with or without the presence of 1% DMSO could be observed.
Furthermore, DSF was used to investigate interactions between ABHD2 (L33-E425) and progesterone or CBK600192, which were described previously.2,3 For this, the protein was prepared as described with the addition of 40 µM of the respective compound and incubated on ice for 30 min prior to centrifugation. Presence of progesterone in solution was monitored by measuring absorbance at 248 nm for different concentrations in sample buffer and the sample.4 For progesterone no significant shift in melting temperature was observed, indicating no stabilization and thereby interaction between progesterone and ABHD2 (L33-E425). Presence of CBK600192 lead to stabilization of the protein by 4.9 °C, which was also observed after incubation with both progesterone and CBK600192 (each 40 µM), indicating binding of CBK600192 which was not disturbed by progesterone.
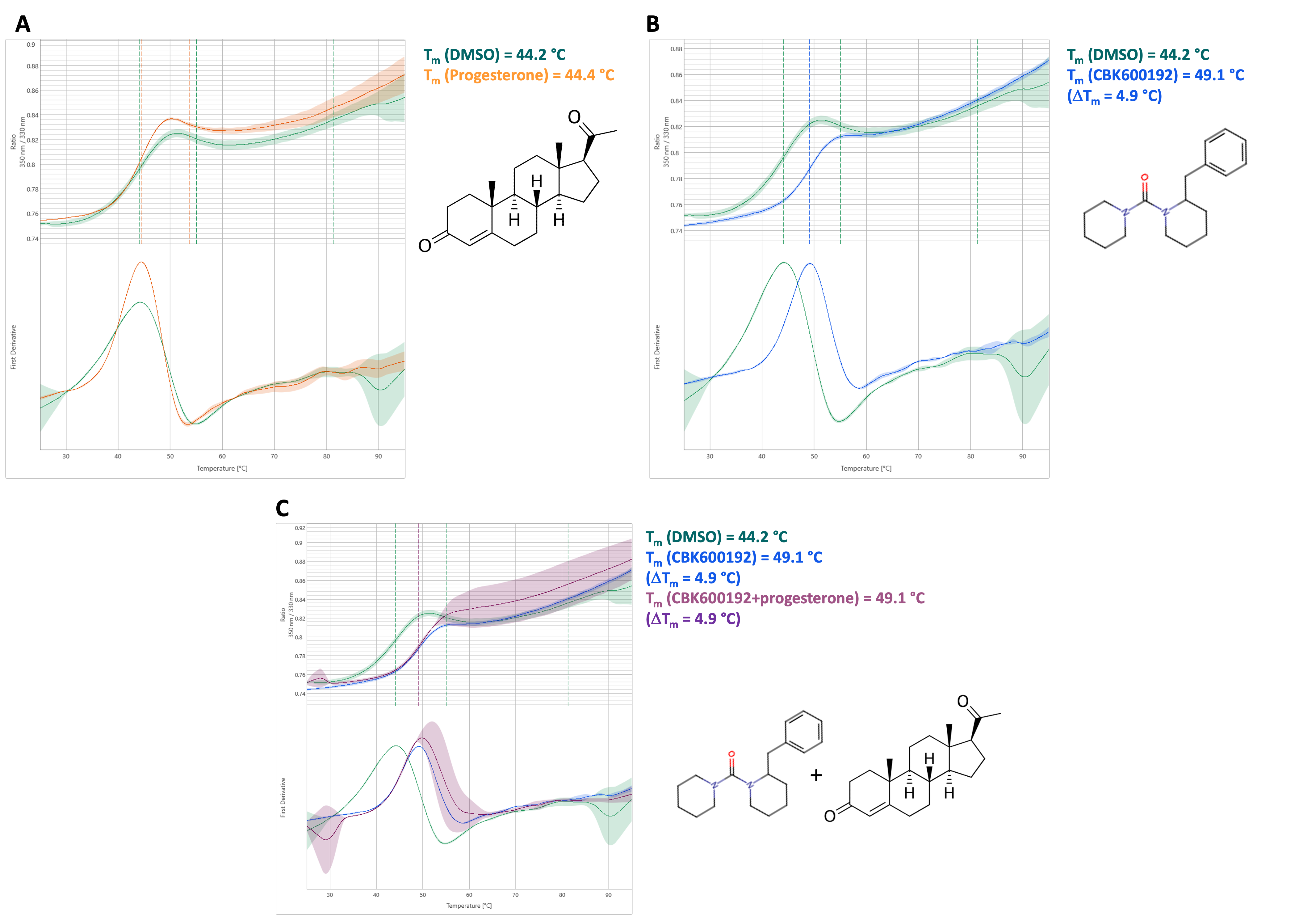
Figure 3: Melting curves of His-FLAG-ABHD2(L33-E425) are displayed in the presence of progesterone, CBK600192, or both in comparison to a DMSO control. 350 nm/330 nm is shown in dependence of temperature [°C].
Activity assay
The more general activity assay for hydrolases that was used for ABHDs 10, 11, and 14B (described here) was adapted to ABHD2. For this, a p-nitrophenyl analogue is incubated with the enzyme, which leads to hydrolysis of the substrate to p-nitrophenol and the respective acid. p-nitrophenol can then be detected due to its absorbance at 405 nm. After initial screens, 50 nM of ABHD2 were incubated with 500 µM p-nitrophenyl octanoate (p-NPO). In contrast to p-nitrophenyl butyrate, which was the substrate used for the other ABHDs, p-NPO was chosen for ABHD2. Not all ABHDs displayed activity towards p-nitrophenyl substrates with longer aliphatic chains, leading to a higher specificity. Additionally, p-NPO displayed higher stability in aqueous solution than analogues with shorter aliphatic chains. Usually, 40 µl of protein solution was pre-incubated with a specific compound on ice for 30 min, transferred to a 96-well plate and then placed in a plate reader with a thermostat function at 37 °C for 2 min. In parallel, buffer was incubated at 37 °C for 30 min, then p-NPO was added to the buffer, mixed, and 160 µl of substrate solution added to each well containing 40 µl protein solution, thereby starting the reaction. This resulted in final assay conditions of 20 mM HEPES, 500 mM NaCl, 10% glycerol, pH 8, 1 mM TCEP, 2% DMSO, 1.44% Methanol, 500 µM p-NPO. Absorbance at 405 nm was typically monitored for 10 min at 37 °C.
Alternatively, a second, fluorescence based, activity assay was developed for ABHD2, using 7-Hydroxycoumarinyl arachidonate (7-HCA) as substrate. As a derivative of arachidonic acid, 7-HCA is structurally very close to ABHD2’s proposed natural substrate 2-arachidonoyl glycerol (2-AG), resulting in a more specific enzyme-substrate interaction.2 7-HCA is hydrolyzed by ABHD2, producing arachidonic acid and umbelliferone, the latter of which can be detected via fluorescence. Also, greater sensitivity due to fluorescence of the product and therefore use of lower concentrations of protein enables determination of lower IC50 values as well as less consumption of protein, which is better suited for high-throughput screens. Solutions were typically prepared as described above, with final concentrations for ABHD2 and 7-HCA of 10 nM and 5 µM, respectively. Solutions were incubated at 37 °C for 10 min and changes in fluorescence monitored (λex = 335 nm, λem = 450 nm).
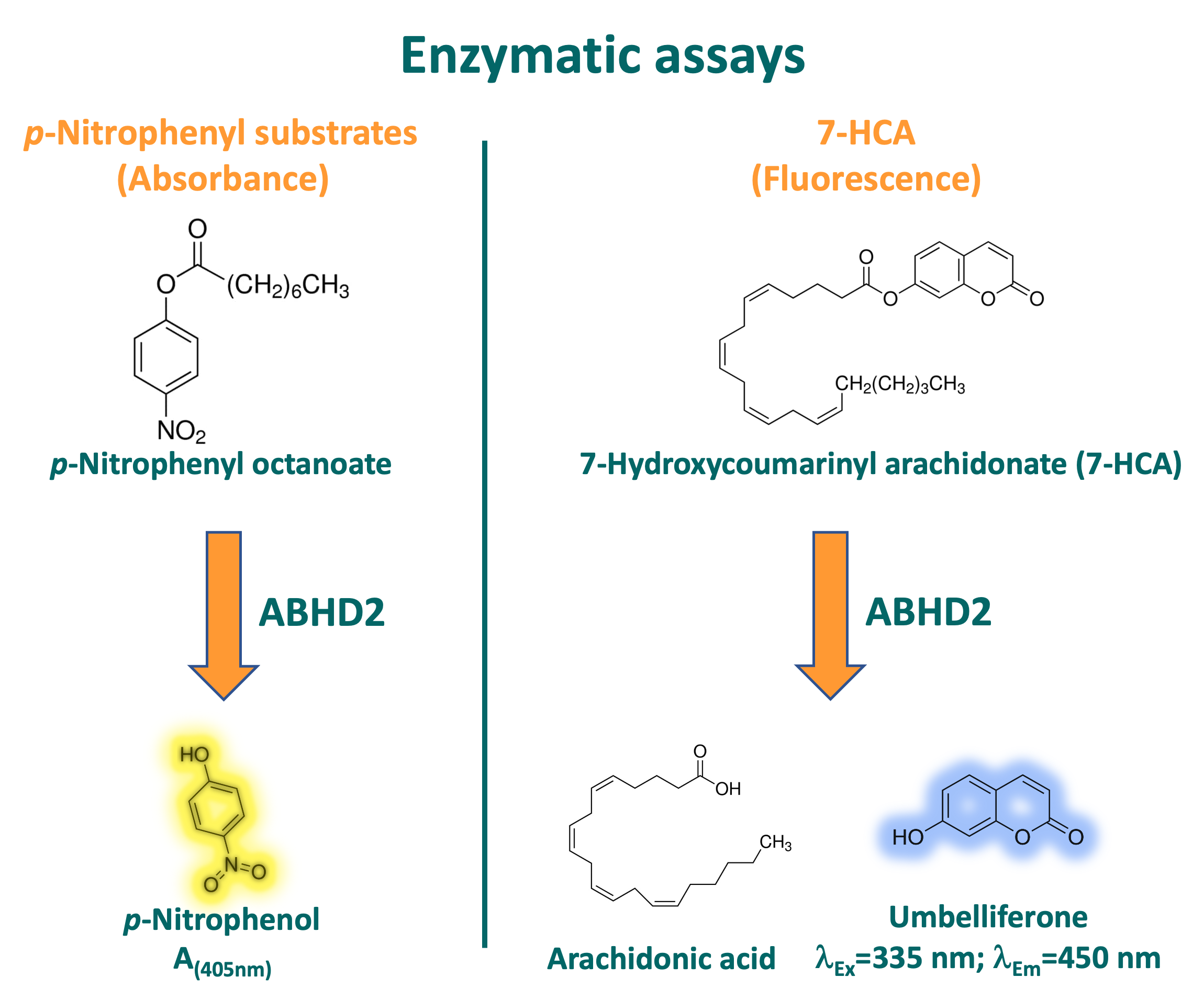
Figure 4: Principle of substrate consumption and mode of detection for both developed activity assays for ABHD2.
Based on the observation that the purified ABHD2 (L33-E425) showed activity in both assays, the effects of progesterone and CBK600192 on the enzymatic activity were investigated. For progesterone, two different concentrations of the ABHD2 were used to be able to observe an increase in activity, however no effect on the enzymatic activity was observed. Contrarily, pre-incubation with CBK600192 lead to a decrease in activity of 87.4% after 10 min of incubation at 37 °C.
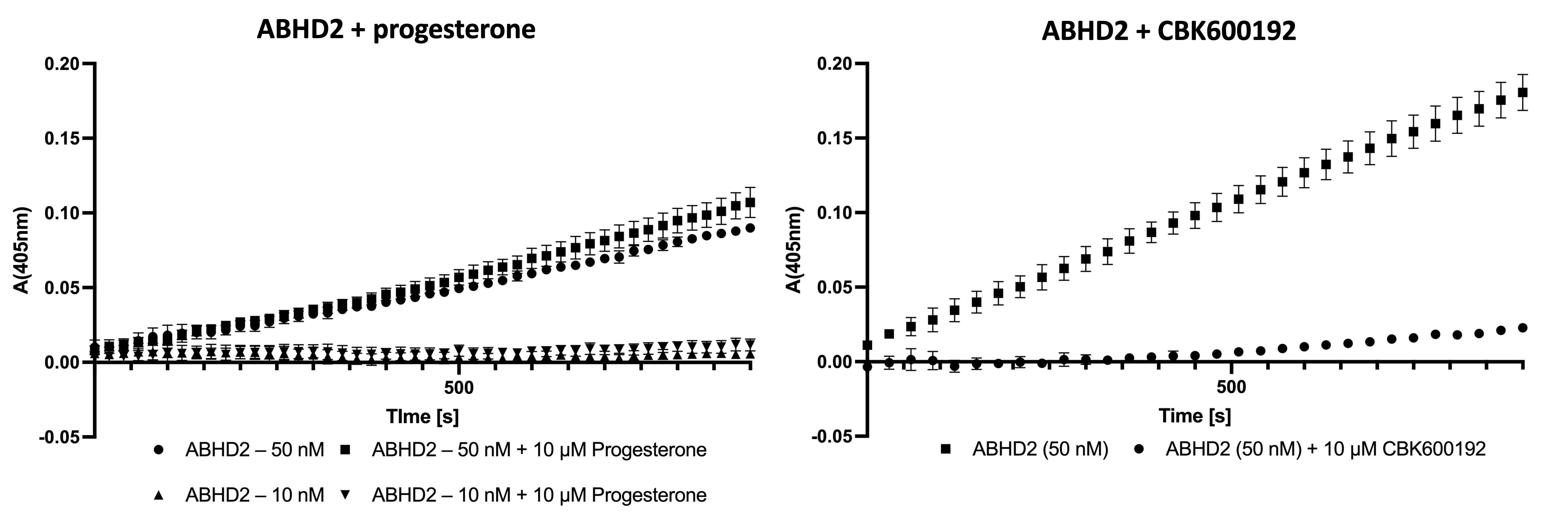
Figure 5: Effect of progesterone (left) and CBK600192 (right) on enzymatic activity of ABHD2. No effect on activity could be observed in the presence of 10 µM progesterone at two different concentrations of ABHD2. Pre-incubation with CBK600192 lead to decrease of activity after 10 min of 87.4%.
Compound screens
Because of the initial results ABHD2 of inhibition by CBK600192, a library consisting of 53 synthesized or commercially available derivatives was collated and screened against at 10 µM in triplicates. The full library can be found in the Zenodo deposition. IC50 values for the eight compounds displaying greatest inhibitory effect relative to the DMSO control.
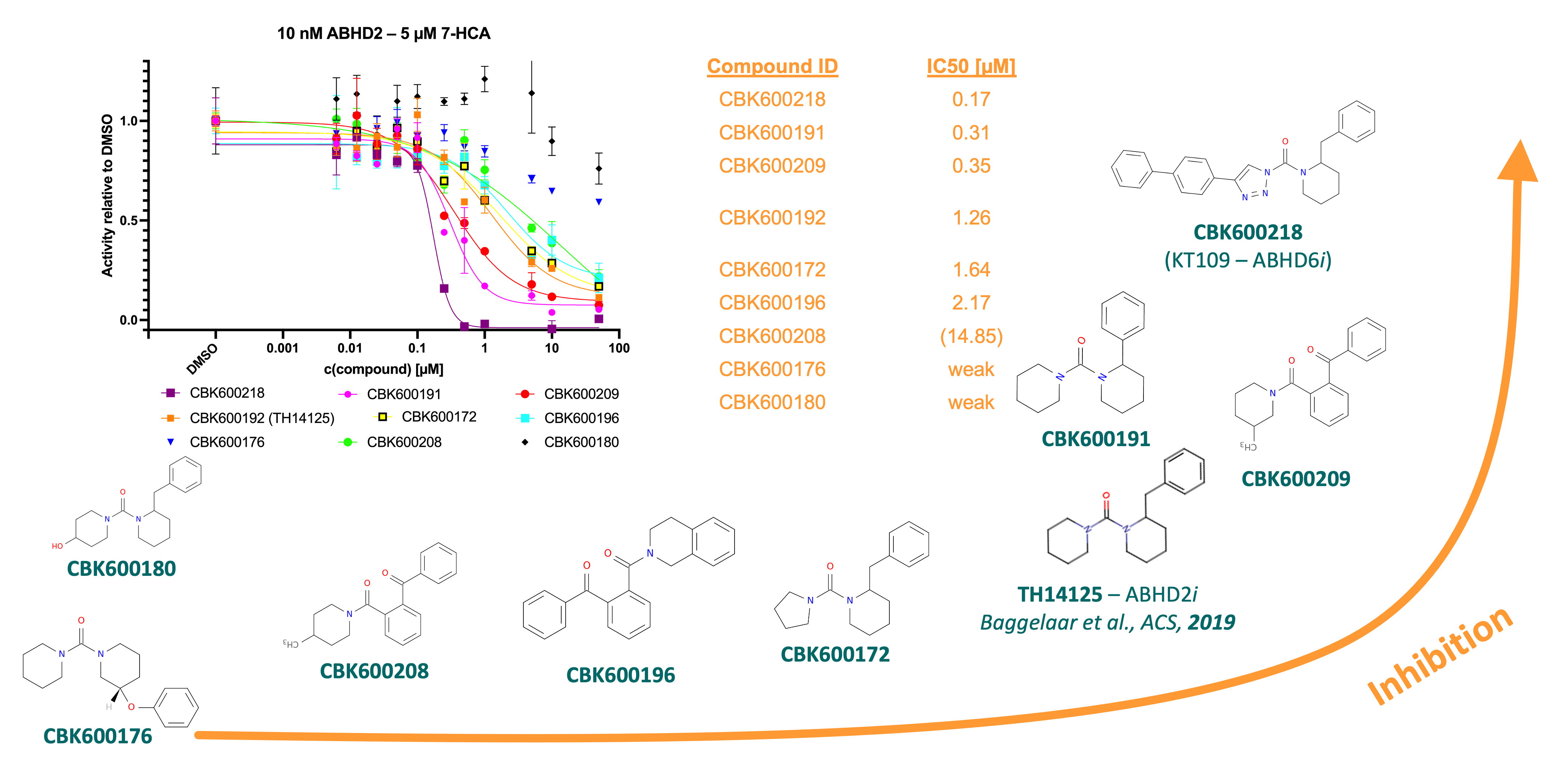
Figure 6: Summary of screening derivatives of published ABHD2 inhibitor CBK600192.3 Three compounds, CBK600218, 191, and 209 showed ~10-fold more potent inhibition in activity assay.
Compounds CBK600218, CBK600191, and CBK600209 displayed a 10-fold greater potency than the published inhibitor found by Baggelaar et al. These were verified by DSF and lead to thermal shifts of 5.8, 13, and 14.4 °C, respectively. The most potent inhibitor, compound 218, also known as KT-109, is a known inhibitor of ABHD6 and proposed to covalently modify the catalytic serine.5 This was investigated using mass spectrometry. Pre-incubation of ABHD2 (L33-E425) with compound 192 did not lead to an increase in mass, however for 218 an increase of 201 g/mol was observed, which is consistent with a covalent modification of the catalytic serine by the mechanism shown below.
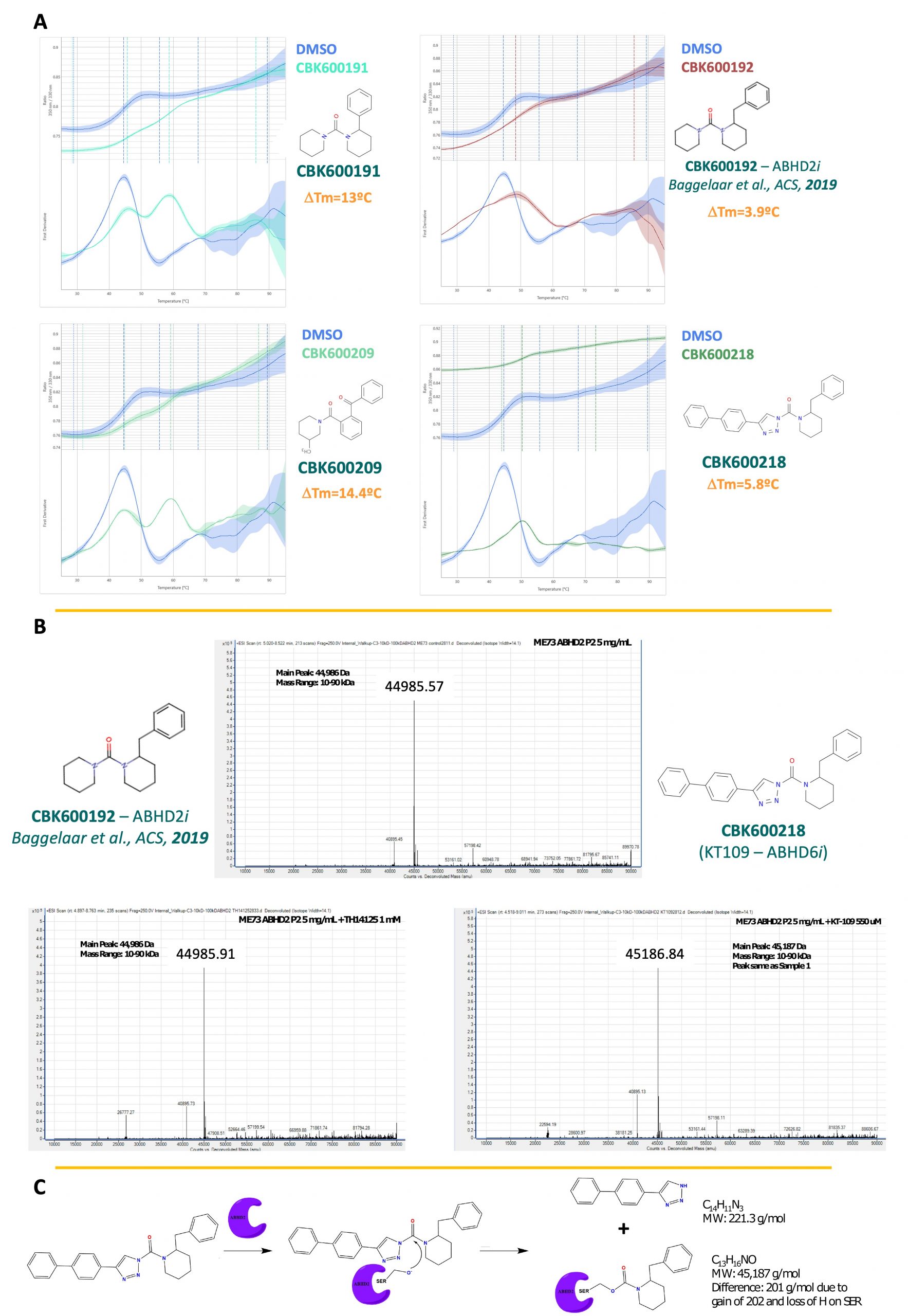
Figure 7: A) CBK600218, 191, and 209 stabilize ABHD2 in DSF. B) Change in molecular weight suggests covalent modification of ABHD2 by CBK600218, which was not observed for CBK600192. C) Proposed mechanism of modification by CBK600218.
Structure
For a complete TEP as well as SAR to develop a more potent and specific inhibitor for ABHD2, a structure of the protein bound or unbound to a small compound is needed. For this, several buffer screens were tested for a construct carrying an N-terminal His-FLAG tag for ABHD2(L33-E425) with or without CBK600192. Several conditions yielded cubic crystals that contained protein according to UV imaging, however none of these crystals diffracted.
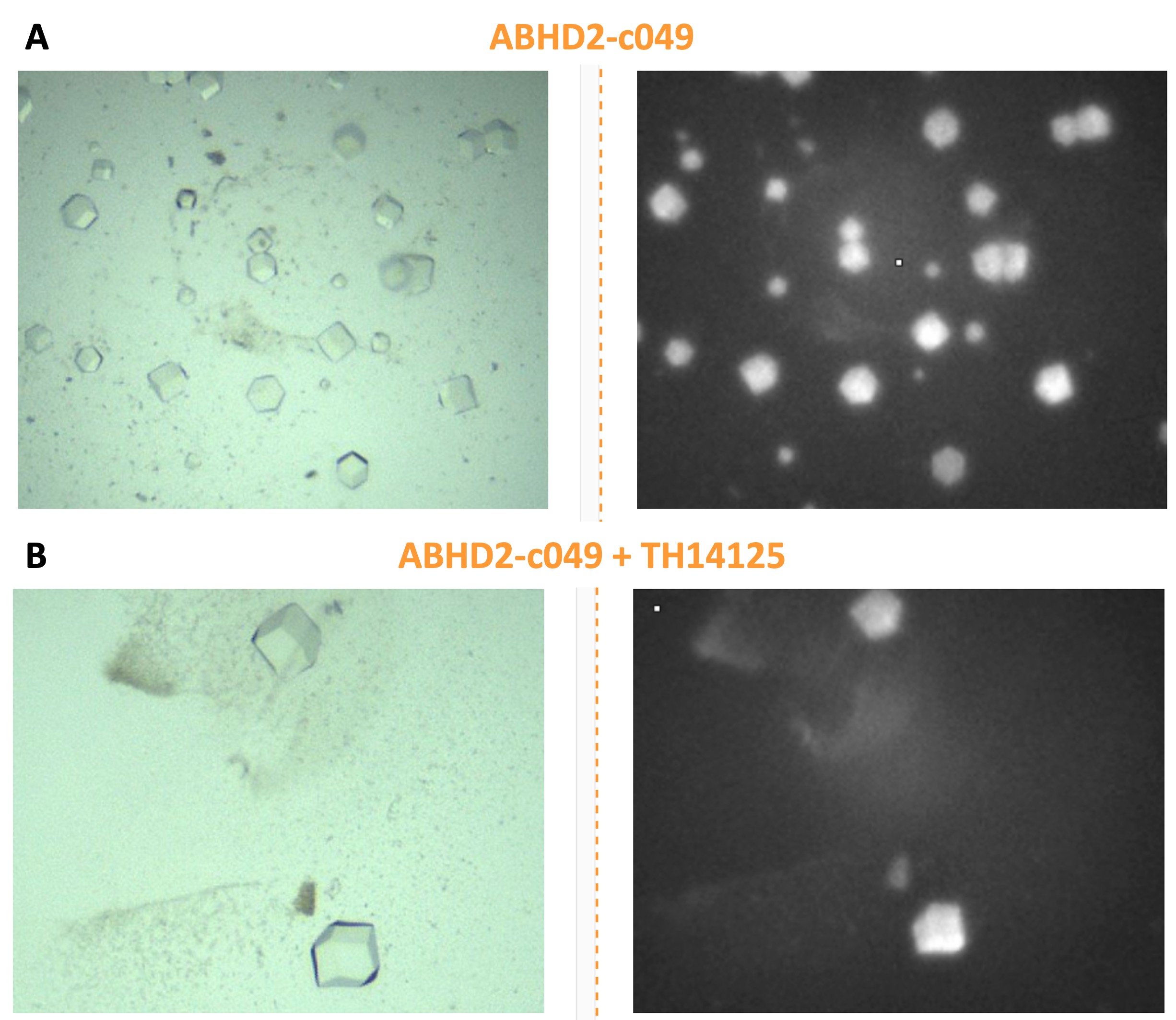
Figure 8: Cubic crystals of ABHD2 incubated with and without the published inhibitor CBK600192. Illumination in UV imaging suggests presence of protein, however none of the crystals diffracted.
Because crystallization of ABHD2 turned out to be challenging, hydrogen-deuterium exchange mass spectrometry (HDXMS) was done in Derek Wilson’s lab at York university to get an initial idea of compound interaction. For this, the protein is incubated with an excess of compound before placed in D2O, proteolytic digestion, and analysis of the peptides by MS, which is then compared to a control without compound. Interaction with a small molecule and/or accompanying structural rearrangements will lead to changes in exchange between amide protons or other polar protons and deuterons of the solvent because of the formation of new hydrogen bonds. These changes in exchange and subsequently mass can be detected and localized by proteolytic digestion and following MS.
For CBK600192, changes in exchange were observed for helices X and Y, which are directly next to the active site, suggesting binding of the inhibitory compound to that region. Still, for effective SAR a better resolution of the binding site (i.e., crystal structure) is needed.
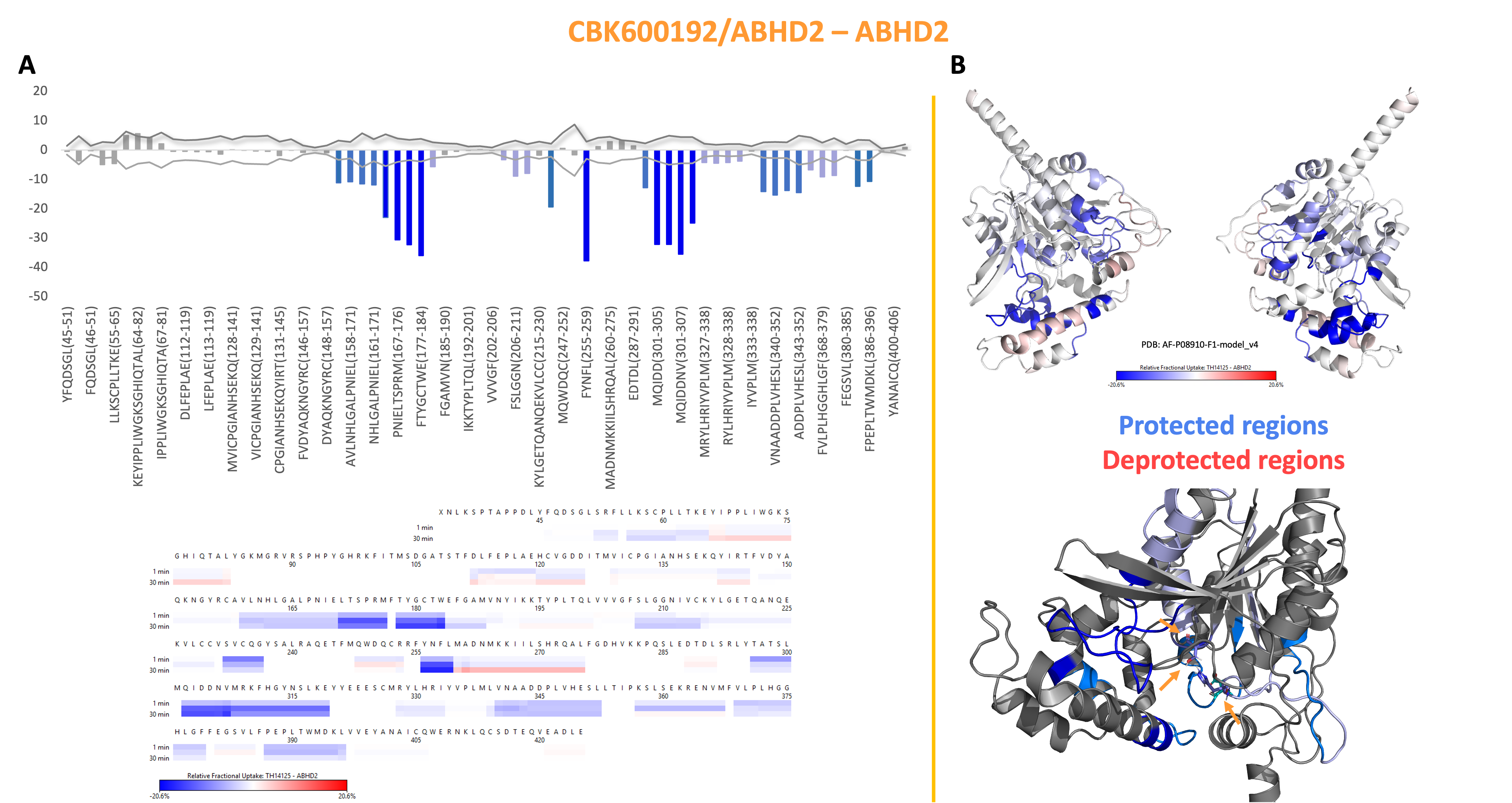
Figure 9: HDXMS data of ABHD2 in presence of CBK600192 revealed changes in H-D exchange close to the active site, suggesting interaction of the inhibitor close by.
Antibodies
At SGC-Montreal currently commercially available antibodies against ABHD2 were tested with lysates from RKO, U-87, and OCUM-1 cell. ABHD2 expression was verified before via RNA sequencing. From these antibodies, none resulted to be specific, producing multiple bands on Western blots, while the most dominant band detected by 14039-1-AP from proteintech correlates to the molecular weight of ABHD2.
In the meantime, ThermoFisher Scientific was contracted to produce specific antibody against ABHD2, based on the recombinantly purified ABHD2 (L33-E425) construct, which is still ongoing.
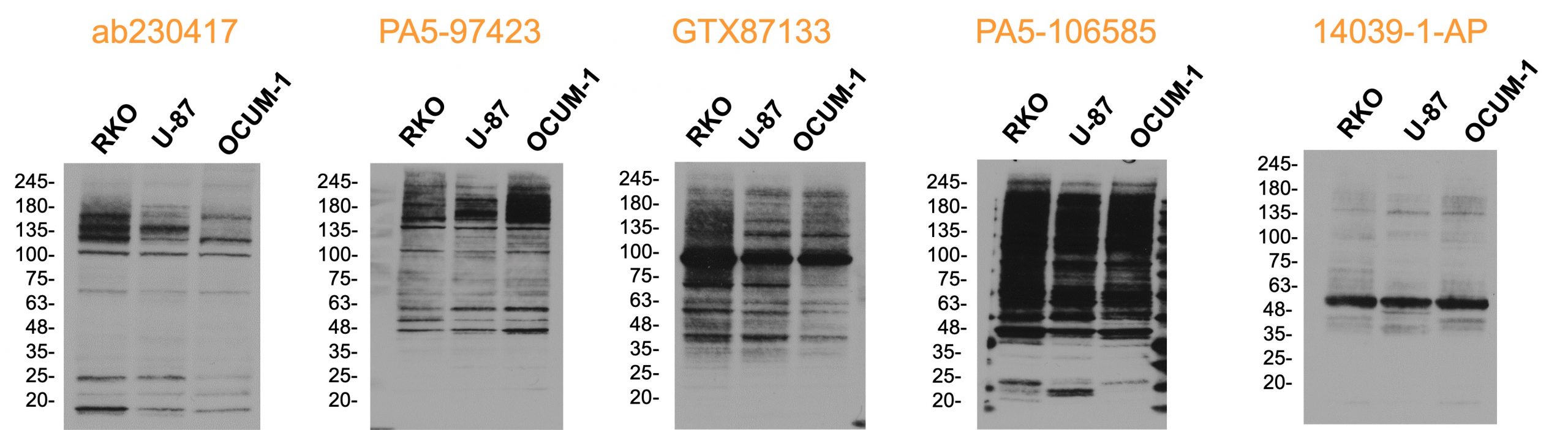
Figure 10: Western blots of different commercially available polyclonal antibodies with lysates of RKO, U-87, and OCUM-1 cells, for which endogenous expression of ABHD2 was verified on RNA level previously.
The Verdict
With all this data, where are we now? We made good progress towards the TEP goal – protocols for protein expression and purification as well as two activity assays are established and tested, also, compounds that are more potent than the published inhibitor CBK600192 have been identified. Antibody generation and efforts towards crystal structures are ongoing, so is the development of a chemical probe. However, the characterization of ABHD2 as a target for non-hormonal contraception led to questions regarding the proposed mechanism. In HEK293T and U2OS cells, no membrane localization of ABHD2 was observed using immunofluorescence or intrinsic eGFP-fluorescence. Moreover, no effect of progesterone on protein stability or activity could be observed, indicating no interaction between progesterone and ABHD2. *Insert passage about hyperactivation assay if Nuvisan is OK with it*
Based on these results, ABHD2 was de-prioritized in a joint meeting between people from SGC, Nuvisan, and the Gates foundation.
The Outlook
What is still missing? After developing a specific antibody against ABHD2, this needs to be validated against a single-knock-out cell line, which is generated at the moment at SGC-Montreal. Afterwards, this antibody can be used to investigate ABHD2 localization in cells with endogenous expression as well as sperm cells. While the results presented here do not support a membrane-bound extracellular domain, overexpression of the protein might lead to non-representative results. Moreover, target engagement of the inhibitory compounds needs to be considered, if ABHD2 is localized inside the cell. Although the compounds are able to inhibit ABHD2, they might not penetrate the plasma membrane. This is explored at SGC Toronto using cellular thermal shaft assays (CETSA) coupled to HiBit as a detection method. Crystal structures of ABHD2 in presence of different ligand will aid the development of more specific compounds. Testing different constructs as well as seeding approaches will hopefully yield diffracting crystals. Finally, the specificity of the identified compounds needs to be validated. For this, a selectivity panel consisting of more ABHDs needs to be developed. Alternatively, CETSA followed by proteomic analysis can be utilized as a more wholistic approach using a cell-line with endogenous ABHD2 expression. The chemical space of the identified compounds will be explored, and a small collection has already been selected. In this context, we are currently starting a collaboration with Recursion. They are specialized in generating compounds libraries guided by machine learning and will provide more compounds to be tested if the current hits are not well suited as chemical probes.
Acknowledgements
All the work presented here is based on the amazing effort of multiple people across the SGC as well as partners from the industry. I would like to thank Aled Edwards, Claudia Tredup, Opher Gileadi, and Daniel Goldberg for scientific guidance and discussions. Madison Edwards, Alma Seitova, Peter Loppnau, and everyone else at SGC Toronto that worked diligently on the production of ABHD2, as well as facilitating experiments conducted at SGC Toronto. Furthermore, I’m grateful for the help of Dalia Barsyte-Lovejoy and her lab at SGC Toronto, especially Michelle Cao and Magdalena Szewczyk, with immunofluorescence and CETSA-HiBit experiments. Likewise, I thank Susanne Müller-Knapp, Yufeng Pan, and Amelie Tjaden at SGC Frankfurt, for their help on cell-based assays, too. On the chemistry side, I appreciate the input and efforts of Matthew Todd and Eve Carter at SGC London, as well as Evert Homan, Martin Haraldsson, and Pauline Ribera at Science for Life Laboratory in Stockholm, likewise the Chemical Biology Consortium Sweden for collating the library and synthesizing compounds. I would like to thank Carl Laflamme, Vincent Francis, and Riham Ayoubi at SGC Montreal for their work on antibody characterization and generation of KO cell lines. Thank you to Martin Moche at the Protein Science Facility of the Karolinksa Institute for his help on crystallization efforts as well as Derek Wilson and Vimanda Chow for their expertise in HDXMS.
This project is funded by the Bill and Melinda Gates Foundation. The Structural Genomics Consortium is a registered charity (no: 1097737) that receives funds from AbbVie, Bayer AG, Boehringer Ingelheim, Genentech, Genome Canada through Ontario Genomics Institute [OGI-196], the EU and EFPIA through the Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer, Takeda and the Wellcome Trust [106169/ZZ14/Z].
References
- Goldenzweig, A. et al. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Mol. Cell 63, 337–346 (2016).
- Miller, M. R. et al. Unconventional endocannabinoid signaling governs sperm activation via the sex hormone progesterone. Science (80-. ). 352, 555–559 (2016).
- Baggelaar, M. P. et al. ABHD2 Inhibitor Identified by Activity-Based Protein Profiling Reduces Acrosome Reaction. ACS Chem. Biol. 14, 2295–2304 (2019).
- Haskins Jr., A. L. Solubility of Progesterone in Water and in Saline. Proc. Soc. Exp. Biol. Med. 70, 228–229 (1949).
- Hsu, K. L. et al. DAGLβ inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat. Chem. Biol. 8, 999–1007 (2012).