Introduction
A current focus in the field of antiviral drug discovery are helicases, which are motor enzymes responsible for ATP-dependent nucleic acid duplex unwinding. Helicases of all superfamilies (SF) have stretches of amino acids called motifs, that are conserved within an SF and similar across SFs. For this reason, established helicase inhibitors interacting with these motifs may inspire the design of lead compounds for other viral helicases. Therefore, we have decided to systematically review the ligands from the ChemBioPort database as a starting point for this process.
A full-screen link for the 3D slides below can be accessed here (DO NOT click on “Download”. Click on “IcmJS”).
RECQL5 Allosteric Ligand (PDB 5LBA)
An allosteric ligand targets human RECQL5, a SF2 helicase belonging to the RecQ subfamily, with diverse functions and roles pertaining to the regulation of DNA repair pathways.
According to previous research, the structure of RECQL5 assumes two distinct conformations, dependent on the presence or absence of ADP. In its Apo form, the N- and C-terminal RecA-like helicase domains are situated in close proximity to one another. In contrast, the helicase domains in the ADP-bound form exhibit a maximal displacement of approximately 30Å from each other (Newman et al., Nucleic Acids Research, 2017). These conformational states of RECQL5 are predicted to be associated with its catalytic cycle, where the Apo form binds to DNA, followed by a transition to its open form upon ATP binding. The open-state conformation triggers ATP hydrolysis and eventually leads to nucleotide unwinding. It is important to note that the ordering and structural intricacies have not yet been comprehensively evaluated (Newman et al., Nucleic Acids Research, 2017).
Since the structural transition of RECQL5 helicase plays a critical role in its activity, inhibition of the conformational change from a closed to an open state is a valid mechanism to impede the activity of RECQL5 helicase. To achieve this inhibition, developing a small molecule that effectively blocks this conformational change is key (Figure 1).
An enzymatic assay from Newman et al. has identified a ligand that targets an allosteric pocket between the N- and C-terminal helicase domains that is only present in the open form of RECQL5. The proposed mechanism for the inhibition of the RECQL5 helicase by a potential allosteric inhibitor is based on the hypothesis that the inhibitor locks the helicase in its open conformation. The locked ADP-bound open form also prevents RNA from binding to the helicase (Newman et al., Nucleic Acids Research, 2017).

Figure 1. An overall crystal structure of RECQL5 with an allosteric inhibitor (PDB 5LBA). The panel on the right is a close-up of the allosteric inhibitor binding site, where key sidechains are depicted as sticks. The allosteric inhibitor is bound in the vicinity of the ADP binding domain, deep at the interface of the N- and C-terminal helicase domain of RECQL5. The proposed mechanism of action for the inhibitor involves locking the RECQL5 helicase in its open conformation, effectively hindering transitional changes linked to the enzyme’s functional activity (Newman et al., Nucleic Acids Research, 2017). Inhibitor: green; ADP yellow; DNA from superimposed structure (2WWY): black; N-terminal helicase domain: orang; C-terminal helicase domain: purple.
Brr2 Allosteric Inhibitor (PDB 5URM and 5URJ)
In previous posts, I analysed two inhibitors targeting the Brr2 helicase, namely an allosteric inhibitor and an RNA-competitive inhibitor (Figure 2).
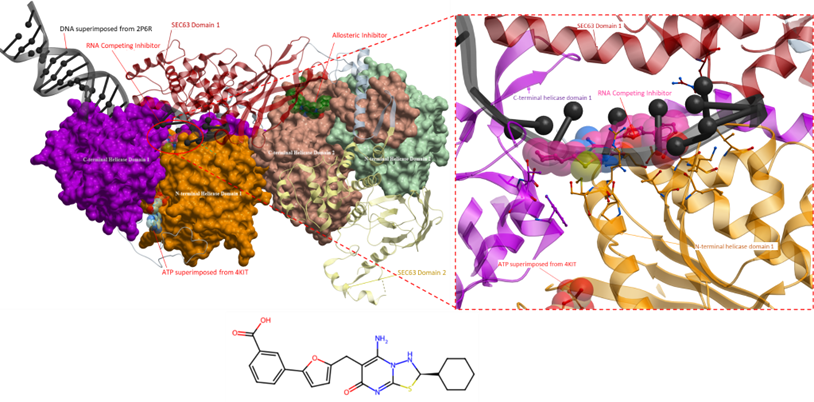
Figure 2. The crystal structure of Brr2 helicase (right) with an RNA competing inhibitor (PDB 5URM; ligand coloured in magenta) and an allosteric inhibitor (PDB 5URJ; ligand coloured in green). The RNA competing inhibitor (right, magenta) occupies the RNA binding site. DNA from superimposed structure (PDB 2P6R) is shown in black; the blue ligands represent ADP from superimposed structure (PDB 4KIT). The N-terminal cassette comprises the N- and C-terminal helicase domains 1 and Sec63 domain 1 (orange, purple and red, respectively). The C-terminal cassette includes the N- and C-terminal helicase domains 2 and Sec63 domain 2 (green, peach and yellow, respectively).
An allosteric inhibitor was found to be more selective than the previously analyzed inhibitors (Iwatani-Yoshihara et al., Journal of Medicinal Chemistry, 2017). The inhibitor binds to an allosteric site formed by the C-terminal RecA-like helicase domain 2 and Sec63 domain 1 (Figure 3). The inhibition mechanism of this allosteric ligand is unknown.
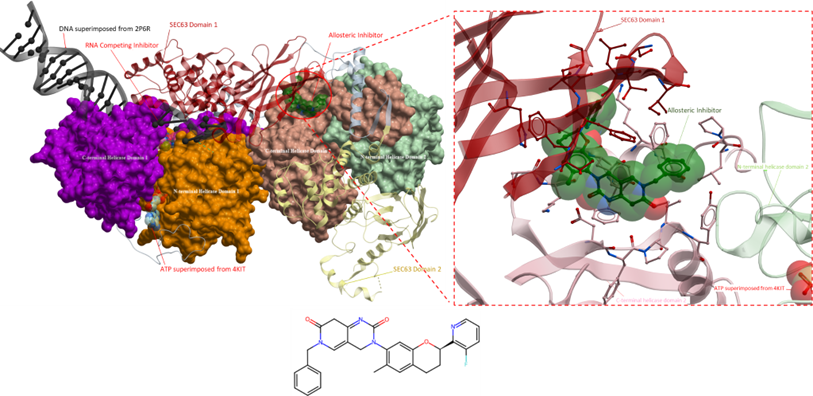
Figure 3. The crystal structure of Brr2 helicase, as shown in Figure 2. Displayed on the right is a magnified view of the binding site of the allosteric inhibitor. The inhibitor binds to a hydrophobic pocket that is at the interface of the N- and C-terminal cassette. However, its mechanism of action is unknown.
Due to the binding of the inhibitor to the Sec63 domain, akin to the allosteric inhibitor from PDB 5URM, it is improbable that this ligand can be chemically repurposed for other helicases, given that the Sec63 domain is not found in any viral helicases and is present only in one other human helicase. Further assessment of the RNA competitive inhibitor is warranted as it has the potential to bind to other SF2 helicases, such as the Dengue viral helicase.
SMARCA2 Allosteric Inhibitor (PDB 6EG3 & 6EG2)
Continuing our analysis of helicase inhibitors, the next compounds under consideration are found to target DNA-dependent helicases of the Snf2 subfamily. Specifically, the SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin subfamily A member 2 (SMARCA2) protein that is involved in chromatin-remodelling complexes and hence involved in the transcriptional regulation of gene expression (Liu et al., Nature, 2017). Despite SMARCA2 being a part of the SF2 helicases, like other helicases from the Snf2 subfamily, they do not have nucleotide unwinding activity. However, they are still considered helicases since helicases are considered ATP-dependent enzymes that can manipulate nucleic acid structures in various ways and are not solely limited to enzymes with duplex unwinding activity (Liu et al., Nature, 2017). SMARCA2 has multiple domains that contribute towards its function including a catalytic ATPase domain that directs chromatin-remodelling and a bromodomain that interacts with acetylated histones on nucleosomes (Papillon et al., Journal of Medicinal Chemistry, 2018). Both domains are targeted by small molecules, however, we will only be discussing the inhibitors that bind to the RecA-like helicase domain.
Both compounds bind to an allosteric pocket within the N-terminal of the RecA-like helicase domain (Figure 4). The compound from PDB 6EG3, J7G, had a mechanism of inhibition that traps the catalytic glutamate (Glu852), from the helicase, in an out conformation via a bidentate interaction with its urea (N-H) (Papillon et al., Journal of Medicinal Chemistry, 2018). The out conformation is believed to be an inactive state of the helicase that is necessary for ATP binding and hydrolysis to occur. Similar to the allosteric inhibitor from 6EG3, the inhibitor from 6EG2 (J7J), the amine on the chloropyridine forms a hydrogen bond with Glu852 suggesting that this compound also inhibits the activity of the SMARCA2 helicase by trapping the catalytic glutamate in an out conformation. The flexibility of the ARL loop (Glu852–His860) also suggests that the two compounds can bind to the helicase in different orientations. The different orientations of the phenylacetylene group in both allosteric inhibitors may affect the movement of helix 6, which could have implications for the helicase’s ATPase activity and ultimately its helicase function (Papillon et al., Journal of Medicinal Chemistry, 2018).
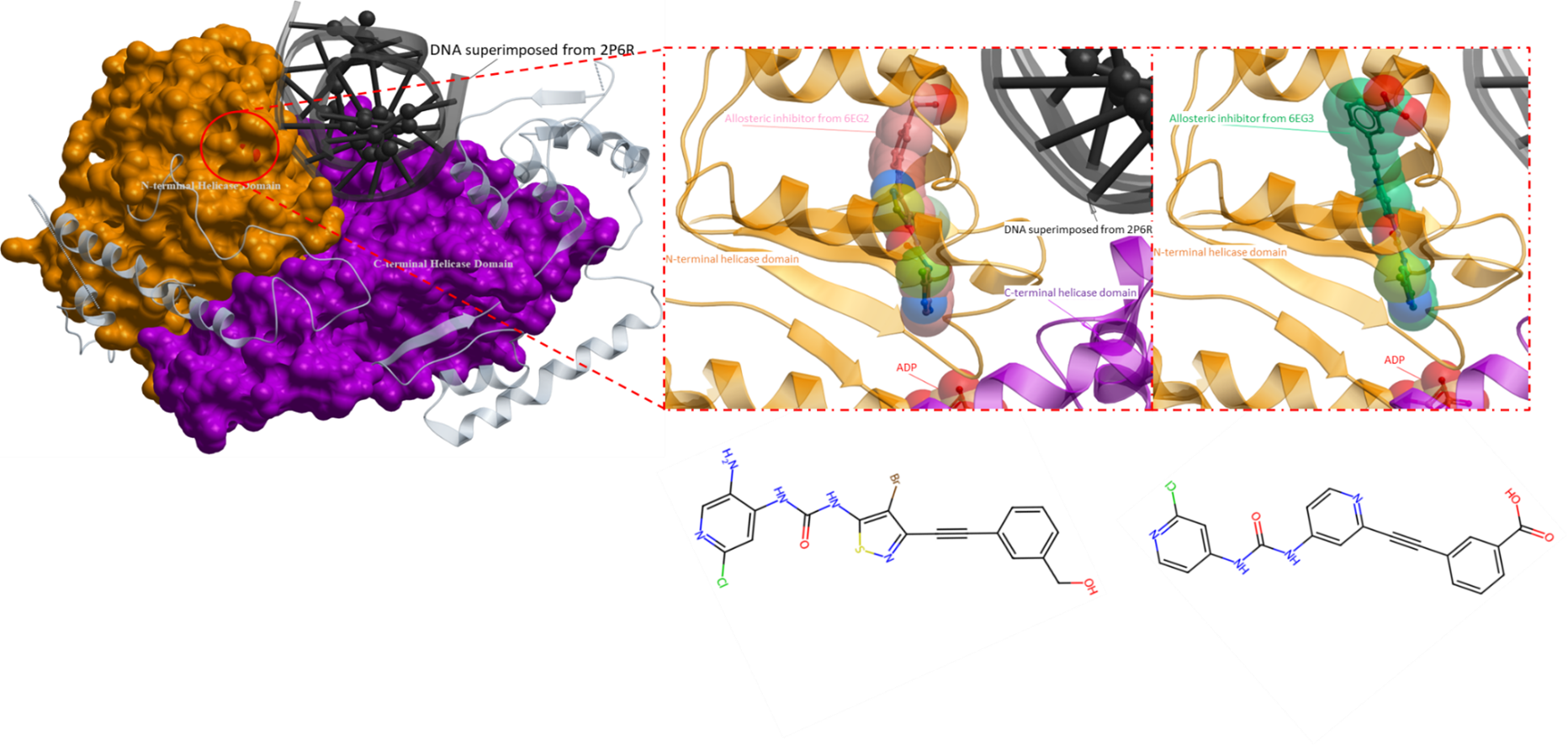
Figure 4. The crystal structure of SMARCA4 (PDB 7VDT) with superimposed allosteric inhibitors (PDB 6EG3 and 6EG2). Both allosteric inhibitors bind at the same allosteric pocket near the ADP binding region (depicted on the right). The definitive mechanism of action of both inhibitors are unknown, however, two speculated mechanisms have been proposed. Firstly, it is hypothesized that the inhibitors induce an outward conformational shift of a catalytic glutamate (Glu852) which hinders ATP hydrolysis and consequently helicase activity. Secondly, the inhibitors are predicted to cause significant movement of helix 6, potentially impacting the helicase’s regular functioning, thus interfering with its activity (Papillon et al., Journal of Medicinal Chemistry, 2018). DNA from a superimposed structure (PDB 2P6R) is shown in black, the pink ligand is the allosteric inhibitor from the PDB structure 6EG2 and the ligand in green is the inhibitor from PDB 6EG3. The N-terminal helicase domain is in orange and the C-terminal helicase domain is in purple.
It is important to note that these allosteric inhibitors not only target SMARCA2 but also its homologue, SMARCA4, which exhibits a high protein-sequence homology of 72% identity, with a 92% identity within their RecA-like helicase domains. The inhibitors are not significantly selective for one helicase over the other and can inhibit both with similar potency (Papillon et al., Journal of Medicinal Chemistry, 2018).
BLM Allosteric Inhibitor (PDB 7AUD)
The next ligand targets the Bloom syndrome protein (BLM) which is also a part of the RecQ family of helicases. BLM is an ATP-dependent helicase that plays a crucial role in maintaining genomic stability and proper DNA repair processes (Chen et al., Elife, 2021). Mutations in the BLM gene have been linked to Bloom syndrome, a rare genetic disorder characterized by DNA repair defects and an increased risk of cancer (Chen et al., Elife, 2021). Therefore, BLM is currently considered a potential target for the development of small molecules that can serve as cancer therapies. In this section, an analysis will be presented on a promising compound that has demonstrated inhibitory activity against BLM helicase function.
The crystal structure (PDB 7AUD) reveals that the ligand does binds to a pocket situated between the N- and C-terminal RecA-like helicase domains, but distinct from the ATP binding site, indicating that the ligand functions as an allosteric inhibitor (Figure 5). The presence of ADP in the crystal structure of BLM, suggests that the helicase is in a state that can bind to ATP and accommodate its hydrolysis. Therefore, the enzyme is in a position that is prepared for the next step in its catalytic cycle, however, the binding of the ligand prevents the conformational changes necessary for further progression (Chen et al., Elife, 2021). This hypothesis is further strengthened by the fact that the allosteric binding site contains a conserved polar bond between Gln975 and His798, which is required for ATPase activity, and is broken once the allosteric inhibitor is bound (Chen et al., Elife, 2021).
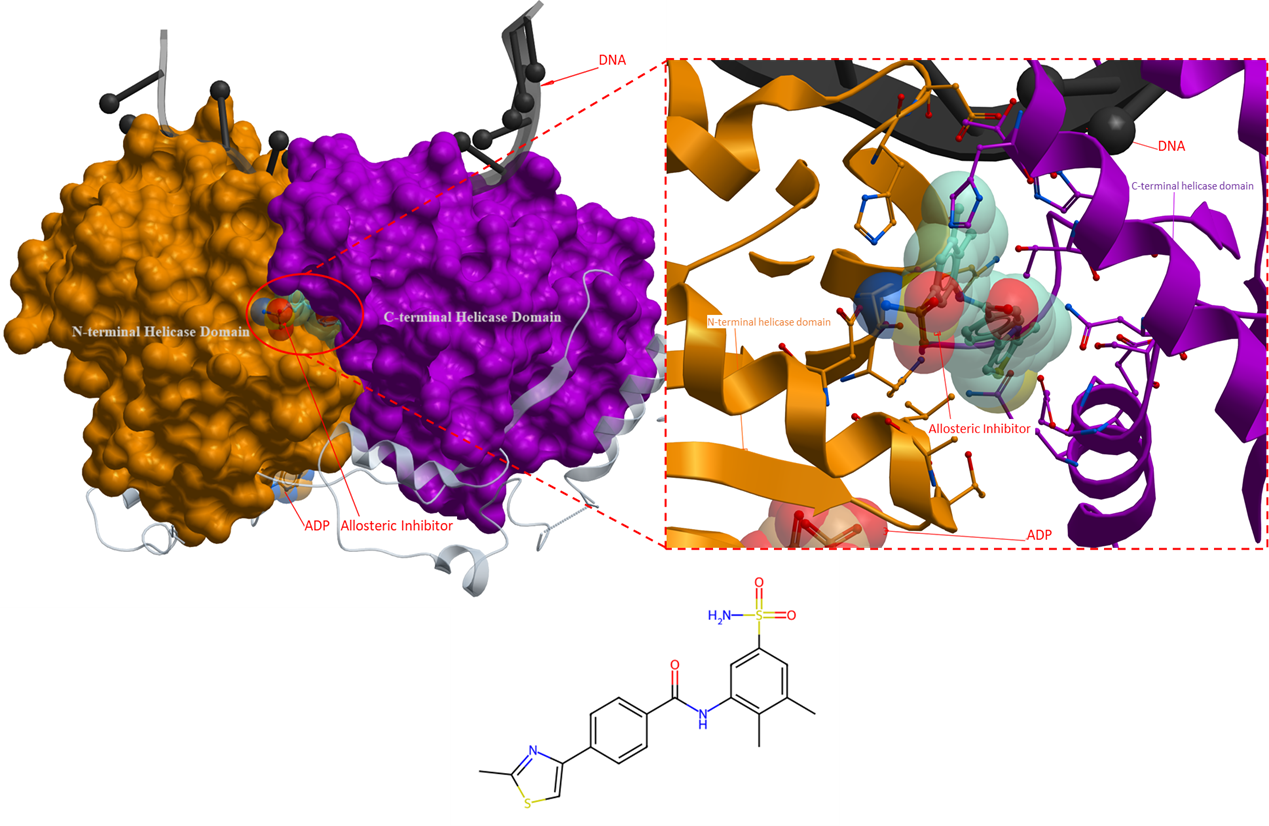
Figure 5. The crystal structure of Bloom syndrome protein with an allosteric inhibitor (PDB 7AUD). The inhibitor binds to a pocket at the interface of the N- and C-terminal helicase domain that locks the helicase in a state unable to progress in its catalytic cycle (Chen et al., Elife, 2021). Allosteric inhibitor: turquoise; ADP: orange. DNA: black. The N- and C-terminal helicase domains are coloured as orange and purple, respectively.
For the allosteric inhibitor to bind to the allosteric pocket in the RecA-like helicase domain, it has been hypothesized that the aromatic-rich loop (ARL; a highly conserved motif in RecQ helicases) must be in a particular conformation (Chen et al., Elife, 2021). In the apo state, the side chain of ARL occupies and obstruct the allosteric binding site. Only when a single-stranded DNA binds to the helicase, does the ARL shift out of the way and create an opening for the allosteric inhibitor to bind to the helicase. This accounts for the finding that the compound is only detectable in the presence of DNA. However, the opposite is also possible, where the binding of the inhibitor results in the rearrangement of the ARL position which then allows the helicase to interact with DNA (Chen et al., Elife, 2021). Kinetic studies are needed to determine if the allosteric inhibitor uses a passive or induced mode of binding to the allosteric binding site.
The selectivity of this ligand to BLM is surprisingly high, despite the fact that the amino acid sequence identity of helicases within the RecQ family is high. It is predicted that the selectivity of the ligand for BLM may be attributed to the significant divergence in the length and amino acid composition of a loop in the ‘Helical Hairpin’ within the RecA-like domain across the RecQ family (Chen et al., Elife, 2021).
Interfacial Inhibitors of EIF4A (PDB 5ZC9 and 6XKI)
The following ligands are interfacial inhibitors of eukaryotic initiation factor 4A (eIF4A). eIFA4 enzymes are categorized as a member of the DEAD-box RNA helicase family, they are involved in many aspects of RNA metabolism (Iwasaki, Molecular Cell, 2019). Two interfacial inhibitors have the same mechanism of action, where the inhibitors interact directly with eIF4A and RNA, and can prevent RNA release (Figure 6 and 7). This method of inhibition prevents ribosomes from scanning mRNAs and thus interferes with translation (Naineni et al., Cell Chemical Biology, 2021).
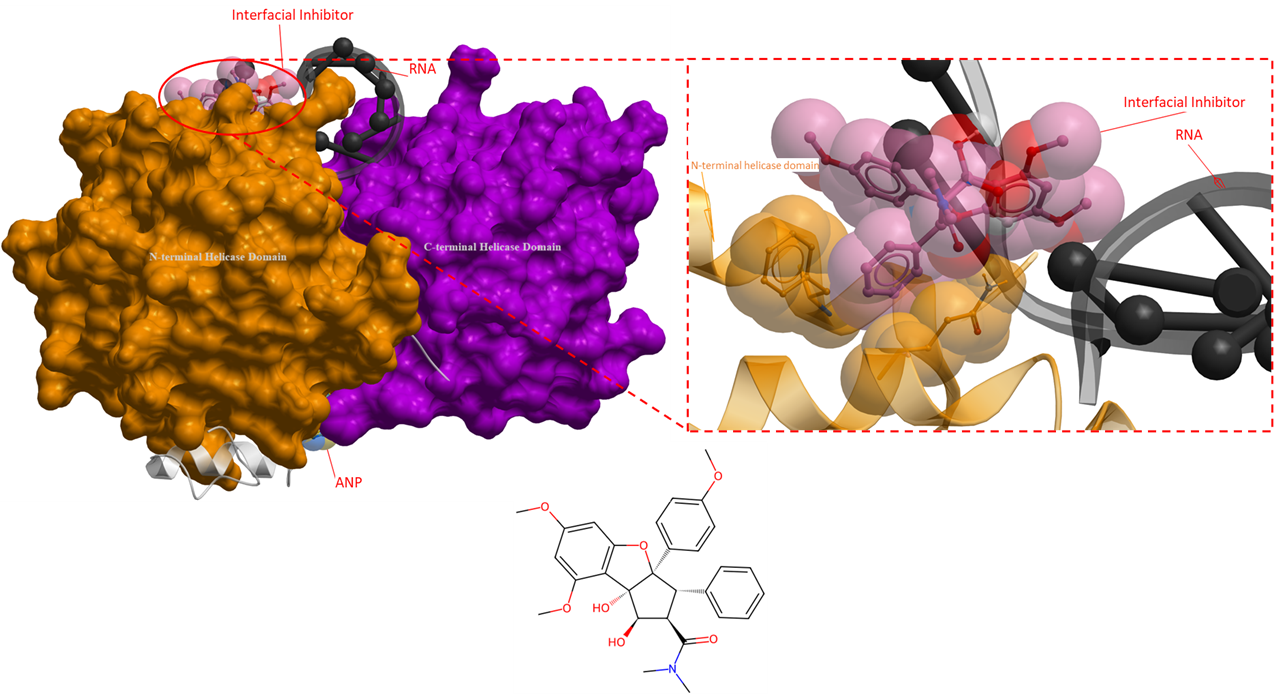
Figure 6. The crystal structure of human eIF4A with an interfacial inhibitor (PDB 5ZC9). This interfacial inhibitor clamps onto both the RNA and eIF4A protein to inhibit protein synthesis (Iwasaki, Molecular Cell, 2019). The interfacial inhibitor is shown in pink, ANP is in yellow, and RNA is black. The N- and C-terminal helicase domains are shown as orange and purple mesh, respectively.
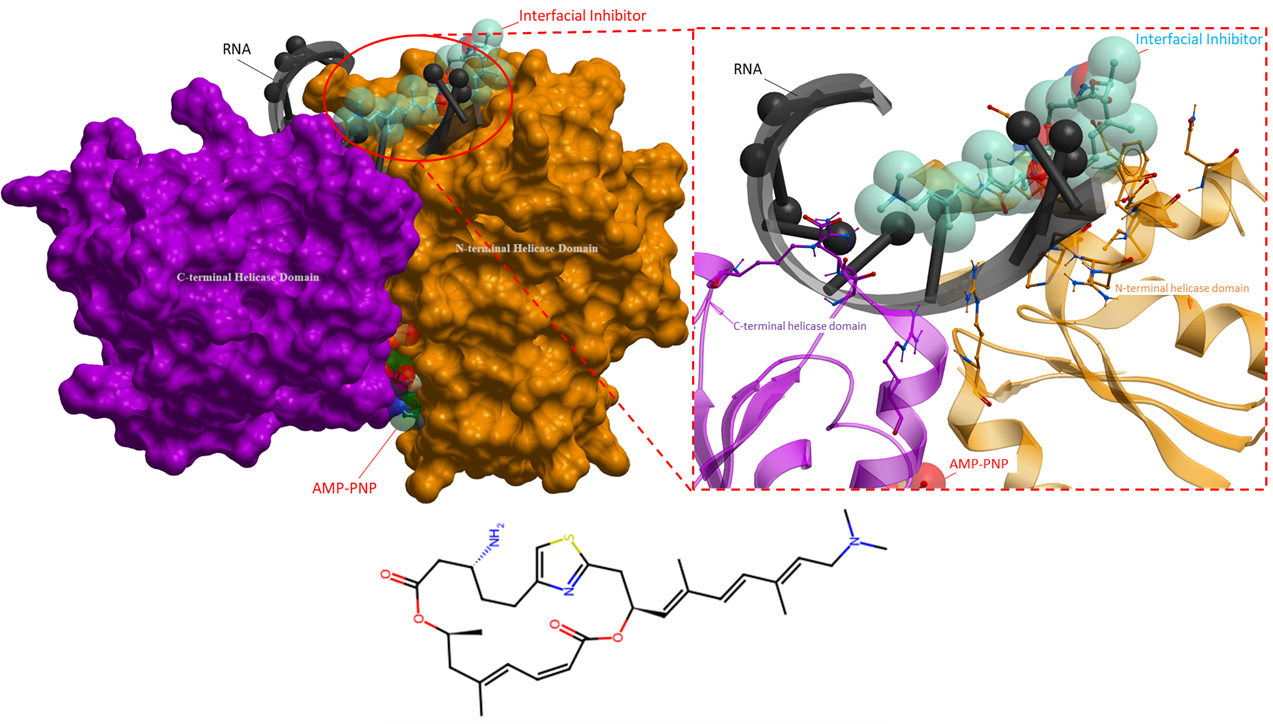
Figure 7. The crystal structure of eIF4A in complex with an interfacial inhibitor (PDB 6XKI). Similarly, to Figure 6, this ligand binds with both the helicase and RNA to prevent translation initiation (Naineni et al., Cell Chemical Biology, 2021). The interfacial inhibitor is blue, ATP analogue is green and RNA black. The N- and C-terminal helicase domains are shown as orange and purple mesh, respectively.
References
Chen, X., Ali, Y. I., Fisher, C. E., Arribas-Bosacoma, R., Rajasekaran, M. B., Williams, G., Walker, S., Booth, J. R., Hudson, J. J., Roe, S. M., Pearl, L. H., Ward, S. E., Pearl, F. M., & Oliver, A. W. (2021). Uncovering an allosteric mode of action for a selective inhibitor of human bloom syndrome protein. eLife, 10. https://doi.org/10.7554/elife.65339
Cox, R. L., Hofley, C. M., Tatapudy, P., Patel, R. K., Dayani, Y., Betcher, M., & LaRocque, J. R. (2019). Functional conservation of RecQ helicase BLM between humans and drosophila melanogaster. Scientific Reports, 9(1). https://doi.org/10.1038/s41598-019-54101-5
Croteau, D. L., Popuri, V., Opresko, P. L., & Bohr, V. A. (2014). Human Recq helicases in DNA repair, recombination, and replication. Annual Review of Biochemistry, 83(1), 519–552. https://doi.org/10.1146/annurev-biochem-060713-035428
Iwasaki, S., Iwasaki, W., Takahashi, M., Sakamoto, A., Watanabe, C., Shichino, Y., Floor, S. N., Fujiwara, K., Mito, M., Dodo, K., Sodeoka, M., Imataka, H., Honma, T., Fukuzawa, K., Ito, T., & Ingolia, N. T. (2019). The translation inhibitor rocaglamide targets a bimolecular cavity between eIF4A and polypurine RNA. Molecular Cell, 73(4). https://doi.org/10.1016/j.molcel.2018.11.026
Iwatani-Yoshihara, M., Ito, M., Klein, M. G., Yamamoto, T., Yonemori, K., Tanaka, T., Miwa, M., Morishita, D., Endo, S., Tjhen, R., Qin, L., Nakanishi, A., Maezaki, H., & Kawamoto, T. (2017). Discovery of allosteric inhibitors targeting the spliceosomal RNA helicase BRR2. Journal of Medicinal Chemistry, 60(13), 5759–5771. https://doi.org/10.1021/acs.jmedchem.7b00461
Liu, X., Li, M., Xia, X., Li, X., & Chen, Z. (2017). Mechanism of chromatin remodelling revealed by the SNF2-nucleosome structure. Nature, 544(7651), 440–445. https://doi.org/10.1038/nature22036
Naineni, S. K., Liang, J., Hull, K., Cencic, R., Zhu, M., Northcote, P., Teesdale-Spittle, P., Romo, D., Nagar, B., & Pelletier, J. (2021). Functional mimicry revealed by the crystal structure of an eIF4A:RNA complex bound to the interfacial inhibitor, Desmethyl pateamine a. Cell Chemical Biology, 28(6). https://doi.org/10.1016/j.chembiol.2020.12.006
Newman, J. A., Aitkenhead, H., Savitsky, P., & Gileadi, O. (2016). Human RECQL5 Helicase. Zenodo. https://doi.org/https://doi.org/10.5281/zenodo.1219714
Papillon, J. P., Nakajima, K., Adair, C. D., Hempel, J., Jouk, A. O., Karki, R. G., Mathieu, S., Möbitz, H., Ntaganda, R., Smith, T., Visser, M., Hill, S. E., Hurtado, F. K., Chenail, G., Bhang, H.-E. C., Bric, A., Xiang, K., Bushold, G., Gilbert, T., … Jagani, Z. (2018). Discovery of orally active inhibitors of brahma homolog (BRM)/SMARCA2 atpase activity for the treatment of brahma related gene 1 (BRG1)/SMARCA4-mutant cancers. Journal of Medicinal Chemistry, 61(22), 10155–10172. https://doi.org/10.1021/acs.jmedchem.8b01318
Shadrick, W. R., Ndjomou, J., Kolli, R., Mukherjee, S., Hanson, A. M., & Frick, D. N. (2013). Discovering new medicines targeting helicases: Challenges and recent progress. SLAS Discovery, 18(7), 761–781. https://doi.org/10.1177/1087057113482586
Lakshi,
That’s a beautiful analysis of the known helicase ligands. It’s really interesting that so many of these inhibitors are allosteric and they appear to work by jamming up the motor rather than directly blocking the ATPase or unwindase activities. Do you find any common regions in or between the RecA domains that you think we should target based on your analysis of the X-ray crystal structures?
Tim
Hi Tim,
I have compared the RecA-like domains of the helicases from this analysis and NSP13. I found that the pockets, where the inhibitors bind are, not conserved or similar to the RecA-like domains in NSP13. I will update again once I finish a complete analysis of the helicase domains from different superfamilies.