Diffuse Intrinsic Pontine Glioma (DIPG) is a type of brain tumour in the brainstem which is highly aggressive and occurs in children. Treatment options for DIPG are very limited because these tumours do not respond to the chemotherapy drugs currently available for adult gliomas. Whole genome and exome sequencing identified several frequently mutated genes in DIPG. H3F3A and HIST1H3B which encode for histone 3.3 and 3.1 respectively were mutated in 78% of the DIPG samples. These mutations result in the substitution of lysine 27 of the histone 3 by methionine (H3K27M). Even though numerous copies of histone H3 are present in the genome, a single copy of H3K27M is sufficient to inactivate histone methyl-transferase EZH2 and alter the epigenetic landscape in the cell.
After histone H3, Activin receptor-Like Kinase-2 (ALK2) is the second most frequently mutated protein in DIPG (24%). ALK2 is a type I BMP receptor serine/threonine kinase encoded by the ACVR1 gene. Arginine 206 and glycine 328 of ALK2 are two of the most commonly mutated residues in DIPG (20% and 56% respectively) and they lead to increased ALK2 activity and responsiveness to a non-native ligand, Activin.
The discovery of ALK2 mutations in DIPG presented a great opportunity because the same mutations, when present in the germline are responsible for Fibrodysplasia Ossificans Progressiva (FOP). In FOP, the connective tissues of patients progressively turn into bone, locking their joints in place. Several labs including Alex Bullock’s had spent years studying the structure and function of ALK2 and developing inhibitors for the treatment FOP. These studies for FOP treatment can serve as the foundation for development of DIPG treatment, thereby extraditing the process. Any progress made will be beneficial to both communities.
Prior to my start at the University of Oxford, an aminopyridine-based kinase inhibitor had been identified at the SGC that bound and potently inhibited ALK2. Recently, more of these compounds have been designed and synthesized by the labs of Paul Brennan and Alex Bullock to improve their metabolic stability and penetrance to the central nervous system (both key attributes for a drug targeting DIPG). The initial promise of these early-stage compounds is now being translated into a far larger drug development programme in an open science venture involving scientists across the globe.
The locations of the ALK2 ATP-binding pocket and sites of disease mutations are indicated in the figure below. Since the ALK2 inhibitors occupy the ATP-binding pocket, it is considered that the wild-type ALK2 and DIPG-causing mutants are similarly susceptible to inhibition.
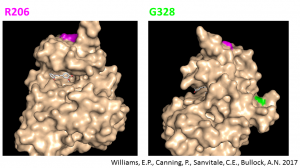
Positions of ALK2 ATP-binding pocket and disease mutations
I am currently establishing several assay systems to determine the cellular IC50 of current and future compounds. They are namely NanoBRET, cellular kinase and dual luciferase-based assays. A NanoBRET target engagement assay will quantify the actual binding of the compounds to ALK2 protein tagged with a NanoLuc luciferase. Before any compound is added, fluorescently labelled tracer which promiscuously binds to the ATP-binding pockets of kinases will accept the light energy emitted by the NanoLuc on ALK2 and experience bioluminescence resonance energy transfer (BRET). The ability of the compounds to out-compete the tracer for the ATP-binding pocket of ALK2 will be measured based on the reduction of the BRET signal. Off-target binding can be controlled for by using other ALKs fused to NanoLuc.
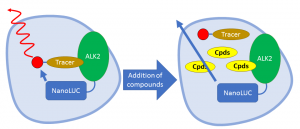
NanoBRET Target Engagement Assay
The cellular kinase assay on the other hand will determine the ability of the compounds to suppress the kinase activity of ALK2 in the cells. When activated by BMP ligands, ALK2 will interact with type II receptors and phosphorylate the transcription factors SMAD1, SMAD5 and SMAD8. In this assay, cells stimulated with BMP ligands in the absence or presence of compounds will be fixed with formaldehyde and stained with antibodies specific to the phosphorylated SMAD proteins. Off-target effects of the compounds on ALK5 can be determined by TGF-β ligand stimulation and by the detection of SMAD2 or SMAD3 phosphorylation. Mutant ALK2 can also be assayed by Activin stimulation and detection of SMAD1/5/8 phosphorylation.
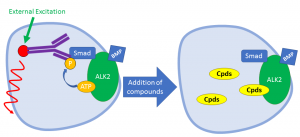
In-cell Western Blot
Currently, a dual luciferase assay has been the most developed assay in the lab. This assay measures the activity of ALK2 indirectly through the transcription activity of the SMAD1/5/8 after they are phosphorylated by ALK2. In this assay, firefly luciferase is expressed under the control of a BMP Response Element (BRE) which can be transcribed when bound by activated SMAD1/5/8. Renilla luciferase is expressed constitutively at low level as a cell number control. When ALK2 is inhibited by the compounds, the amount of firefly luciferase signal will decrease relative to the renilla luciferase signal. In this assay, off-target effects on ALK5 can be determined by using a different construct with firefly luciferase expressed under the control of a CAGA sequence, which is activated by SMAD2/3.
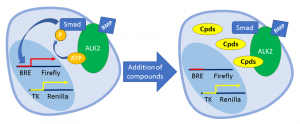
Dual Luciferase Assay
Together, these assays will help guide the selection of the optimal ALK2 inhibitor to take forward into disease models of DIPG and ultimately the development of an investigational drug for the benefit of DIPG patients.
One Reply to “Project overview: Establishing Cellular Assays to Screen for ALK2 Inhibitors”