Tandem Microwave-Facilitated Synthesis of Ether-Containing Oxadiazoles
David A. Rogers and Kevin J. Frankowski
https://doi.org/10.5281/zenodo.6036677
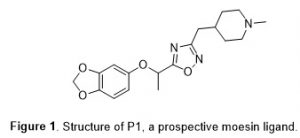
The National Institute on Aging has made a significant commitment to improving the treatment of Alzheimer’s disease through the funding of the TREAT-AD network (Target Enablement to Accelerate Therapy Development for AD, https://treatad.org/). One of the goals of TREAT-AD is to create chemical tools to explore under-studied and unexplored targets related to Alzheimer’s disease. One such target is the protein moesin (MSN), a membrane-organizing extension spike protein.1 We recently identified an oxadiazole-based small molecule, P1 (Figure 1), as a potential ligand for MSN. To support TREAT-AD probe development efforts, we sought to synthesize P1 analogues. Here we describe our synthetic strategy to efficiently construct an initial analogue set of oxadiazoles. The synthetic protocol combines two microwave-facilitated literature methods and should be generally applicable to a wide range of oxadiazole-containing molecules.
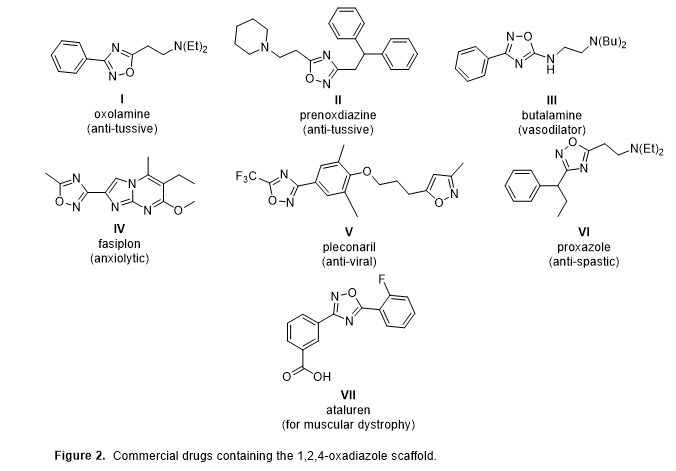
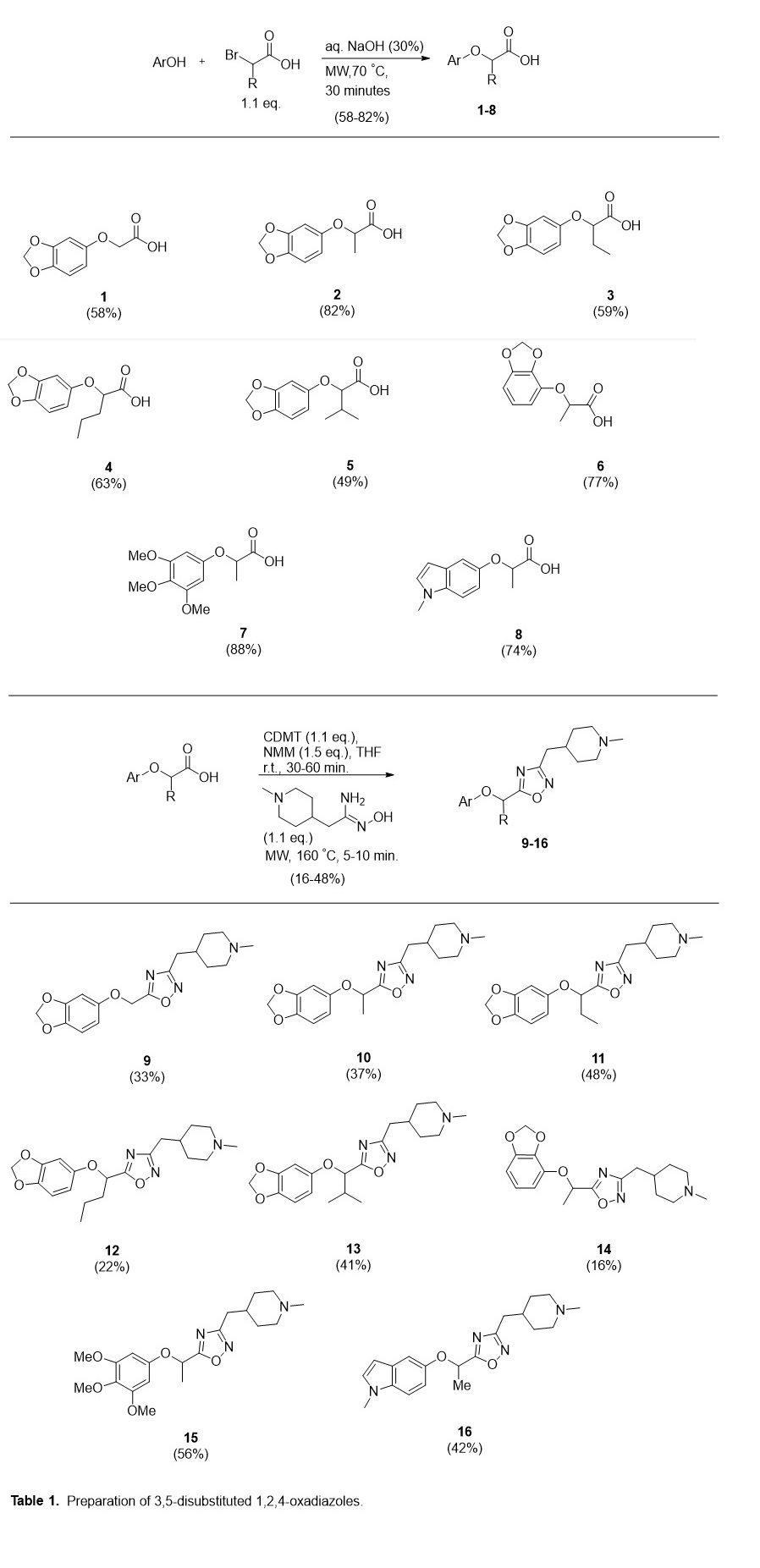
The 3,5-disubstituted 1,2,4-oxadiazole scaffold has demonstrated significant and diverse therapeutic activities, including with several commercial drugs possessing this framework (Figure 2).2 The 1,2,4-oxadiazole framework can function as a bioisostere for amides and esters and generally confers greater metabolic stability and resistance to hydrolysis.3 The synthetic strategy employed in this work couples two separately published microwave-mediated steps to provide the reported products in moderate to good yield while minimizing time investment (Table 1).
We identified a literature protocol for the microwave-mediated SN2 substitution of an α-bromo carboxylic acid with the corresponding phenol in aqueous sodium hydroxide that provides the desired (aryloxy)alkanoic acid in fair to very good yield.4 It is notable that, in this step, the phenol is allowed to fully dissolve in the aqueous NaOH solution before the addition of α-bromo acid and the subsequent microwave irradiation. The dissolution time varied from instantaneous to approximately fifteen minutes, likely depending on the rate of aryl alkoxide formation. Subsequent microwave irradiation for 30–45 minutes achieved 49–88% yields with scale in the 0.25–2.00 mmol range. For some substrates, acidification of the aryloxy(alkanoate) substitution produced the product as a white precipitate which could be recovered by filtration. For cases in which an oil was observed to separate from the aqueous phase, ethyl acetate extraction provided a crude residue separable by chromatography.
The next necessary step to obtain P1 was condensation of the carboxylic acid intermediate with a 1-methylpiperidinyl amidoxime. An amidoxime is often employed with a carboxylic acid activated by common coupling reagents (e.g. DCC, EDC, CDI, TBTU) for the heterocyclization step, though these methods often require long periods of reflux with variable yields. Additionally, many of these procedures are limited to more reactive aryl amidoximes.5 In an approach reported by de Luca et. al., microwave-mediated heterocyclization was achieved after activation of the carboxylic acid using 2-Chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) and N-methylmorpholine. In addition to being compatible with alkyl amidoximes, this microwave-assisted method performed significantly better in comparison studies to conventional heating.6 This strategy was applied to benzodioxol-yl, trimethoxyphenyl, and indol-yl substrates with yields ranging 16–56% (Table 1).
EXPERIMENTAL
General procedure A (preparation of (aryloxy)acetic acids): To a 10 mL microwave reaction vessel equipped with a stir bar was added the phenol (0.5 mmol) and sodium hydroxide (9 M, 0.3 mL, 2.7 mmol). The reaction mixture was allowed to stir at room temperature for 30 minutes. After dropwise addition of α-bromo acid (1.1 mmol), the reaction vessel was sealed and heated under microwave irradiation for 45 minutes at 70 °C. After cooling to room temperature, the pH was adjusted to 1–2 with hydrochloric acid (aqueous, 10 wt. %). The mixture was extracted with ethyl acetate (3 × 10 mL). The combined organic layers were concentrated under vacuum. The resulting residue was purified using C-18 reverse-phase chromatography with acetonitrile and water w/ 0.1% formic acid.
General Procedure B (preparation of oxadiazole analogues): To a 10 mL microwave reaction vessel equipped with a stir bar, was charged the aryl ether intermediate (0.5 mmol) suspended in anhydrous THF (0.75 mL). 2-Chloro-4,6-dimethoxy-1,3,5- triazine (97.8 mg, 0.55 mmol) and N–methylmorpholine (170 µL, 1.55 mmol) were added and the reaction mixture was allowed to stir for 30–60 minutes. (Z)-N’-Hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (88.3mg, 0.52 mmol) and toluene (1 mL) were added. The resulting mixture was heated under microwave irradiation for 5–15 minutes at 160 °C and allowed to cool. The solvent was removed under vacuum and the residue chromatographed directly with C-18 reverse-phase using acetonitrile and water w/ 0.1% NH4OH.
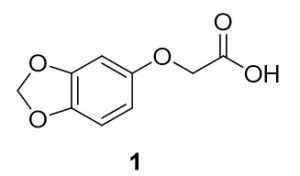
2-(benzo[d][1,3]dioxol-5-yloxy)-2-methylpropanoic acid (1). Sesamol (138.6 mg, 1.00 mmol) and 2-chloroacetic acid (96.8 mg, 1.02 mmol) were reacted according to General Procedure A to furnish the ether 1 as an off-white solid (114.3 mg, 0.58 mmol, 58%). 1H NMR (400 MHz, CDCl3): δ ppm 4.60 (2H, s), 5.94 (2H, s), 6.32–6.35 (1H, dd, J = 2.5, 8.4 Hz), 6.54–6.55 (1H, d, J = 2.7 Hz), 6.70–6.72 (1H, d, J = 8.2 Hz); LC-MS (ESI) m/z: [M+H]+ Calcd for C9H9O5 197.04; found 197.28.
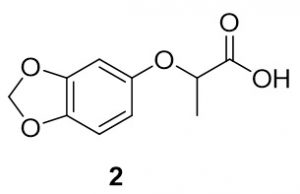
2-(Benzo[d][1,3]dioxol-5-yloxy)propanoic acid (2). Sesamol (278.3 mg, 2.0 mmol) and 2-bromopropionic acid (340mg, 2.2 mmol) were reacted according to General Procedure A to furnish the ether 2 as an off-white solid (345.4 mg, 1.64 mmol, 82%). 1H NMR (400 MHz, DMSO-d6): δ 1.43 (d, J = 6.7 Hz, 3H), 4.69 (q, J = 6.9 Hz, 1H), 5.93 (s, 2H), 6.27 (dd, J = 2.7 Hz, 1H), 6.55 (d, J = 2.4 Hz, 1H), 6.77 (d, J = 8.6 Hz, 1H), 12.87 (bs, 1H); LC-MS (ESI) m/z: [M+H]+ Calcd for C10H11O5 211.06; found 211.29.
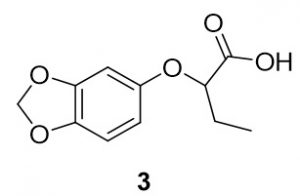
2-(benzo[d][1,3]dioxol-5-yloxy)butanoic acid (3). Benzo[d][1,3]dioxol-4-ol (68.1 mg, 0.49 mmol) and 2-bromobutanoic acid (84.5 mg, 0.51 mmol) were reacted according to General Procedure A to furnish the ether 3 as an off-white solid (65.5 mg, 0.29 mmol, 59%). 1H NMR (400 MHz, methanol-d4): δ ppm 0.86–0.90 (3H, t, J = 7.4 Hz), 1.68–1.81 (2H, m), 4.28–4.31 (1H, dd, J = 5.3, 6.9 Hz), 5.69 (2H, s), 6.14–6.16 (1H, dd, J = 2.5, 8.4 Hz), 6.33 (1H, d, J = 2.4 Hz), 6.48–6.40 (1H, d, J = 8.6 Hz); LC-MS (ESI) m/z: [M+H]+ Calcd for C11H13O5 225.08; found 225.23.
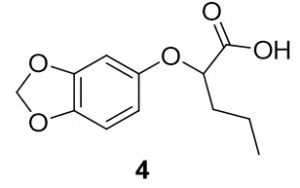
2-(benzo[d][1,3]dioxol-5-yloxy)pentanoic acid (4). Benzo[d][1,3]dioxol-4-ol (70.9 mg, 0.51 mmol) and 2-bromopentanoic acid (101.7 mg, 0.56 mmol) were reacted according to General Procedure A to furnish the ether 4 as an off-white solid (76.4 mg, 0.51 mmol, 63%). 1H NMR (400 MHz, methanol-d4): δ ppm 0.86–0.90 (3H, t, J = 7.4 Hz), 1.40–1.50 (2H, m), 4.43–4.46 (1H, t, J = 6.3 Hz), 5.78 (2H, s), 6.21–6.24 (1H, dd, J = 2.5, 8.4 Hz), 6.40–6.41 (1H, d, J = 2.4 Hz), 6.57–6.59 (1H, d, J = 8.2 Hz); LC-MS (ESI) m/z: [M+H]+ Calcd for C12H15O5 2.09; found 239.32.
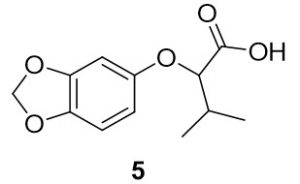
2-(benzo[d][1,3]dioxol-5-yloxy)-3-methylbutanoic acid (5). Benzo[d][1,3]dioxol-4-ol (137.9 mg, 1.00 mmol) and 2-bromo-3-methylbutanoic acid (204.3 mg, 1.13 mmol) were reacted according to General Procedure A to furnish the ether 5 as an off-white solid (116.6 mg, 0.49 mmol, 49%). 1H NMR (400 MHz, methanol-d4): δ ppm 0.86–0.90 (3H, t, J = 7.4 Hz), 1.40–1.50 (2H, m), 1.75–1.81 (2H, m), 4.43–4.46 (1H, t, J = 6.3 Hz), 5.78 (2H, s), 6.21–6.24 (1H, dd, J = 2.5, 8.4 Hz), 6.40–6.41 (1H, d, J = 2.4 Hz), 6.57–6.59 (1H, d, J = 8.2 Hz); LC-MS (ESI) m/z: [M+H]+ Calcd C12H15O5 for 239.09; found 239.16.
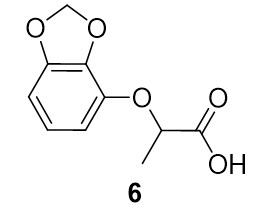
2-(benzo[d][1,3]dioxol-4-yloxy)propanoic acid (6). Benzo[d][1,3]dioxol-4-ol (34.4 mg, 0.25 mmol) and 2-bromopropionic acid (40.4 mg, 0.264 mmol) were reacted according to General Procedure A to furnish the ether 6 as an off-white solid (40.1 mg, 0.19 mmol, 77%). 1H NMR (400 MHz, CDCl3): δ ppm 1.65–1.67 (3H, d, J = 6.7 Hz), 4.95–5.00 (1H, q, J = 6.9 Hz), 5.94–5.96 (2H, m), 6.51–6.58 (2H, m), 6.74–6.78 (1H, t, J = 8.0 Hz); LC-MS (ESI) m/z: [M+H]+ Calcd for C10H11O5 211.06; found 211.48.
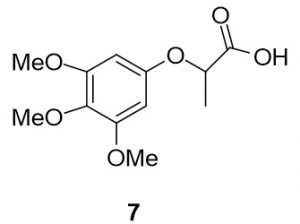
2-(3,4,5-trimethoxyphenoxy)propanoic acid (7). 3,4,5-trimethoxyphenol (183.9 mg, 1.00 mmol) and 2-bromopropionic acid (169.6 mg, 1.11 mmol) were reacted according to General Procedure A to furnish the ether 7 as an off-white solid (225.1 mg, 0.88 mmol, 88%). 1H NMR (400 MHz, CDCl3): δ ppm 1.62–1.63 (3H, d, J = 7.0 Hz), 3.77 (s, 3H), 3.80 (s, 6H), 4.71–4.77 (1H, q, J = 6.7 Hz), 6.15 (s, 2H); LC-MS (ESI) m/z: [M+H]+ Calcd for C12H17O6 257.10; found 257.34.
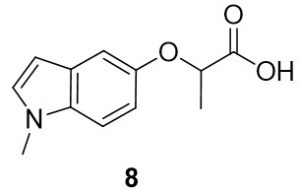
2-((1-methyl-1H-indol-5-yl)oxy)propanoic acid (8). 1-methyl-1H-indol-5-ol (54.4 mg, 0.25 mmol) and 2-bromopropionic acid (47.5 mg, 0.27 mmol) were reacted according to General Procedure A to furnish the ether 8 as an off-white solid (64.8 mg, 0.25 mmol, 74%). 1H NMR (400 MHz, CDCl3): δ ppm 1.65–1.67 (3H, d, J = 6.7 Hz), 3.76 (3H, s), 4.78–4.83 (1H, q, J = 7.0 Hz), 6.40 (1H, d, J = 2.7 Hz), 6.92–6.94 (1H, dd, J = 2.4, 8.6 Hz), 7.03–7.04 (1H, d, J = 2.0 Hz), 7.10–7.11 (1H, d, J = 2.4 Hz), 7.21–7.24 (1H, d, J = 8.6 Hz); LC-MS (ESI) m/z: [M+H]+ Calcd C12H14NO3 for 220.10; found 220.20
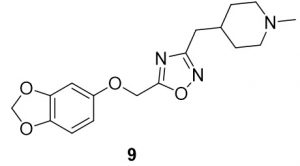
5-(1-(benzo[d][1,3]dioxol-5-yloxy)ethyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (9). Compound 1 (24.1 mg, 0.12 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (23.2 mg, 0.14 mmol) were reacted according to General Procedure B to furnish the oxadiazole 9 as a pale yellow solid (13.5 mg, 0.04 mmol, 33% yield). Rf = 0.42 (10% MeOH in DCM); mp = 65–67 ◦C; 1H NMR (400 MHz, methanol-d4) δ ppm 0.98 (s, 1H), 1.47–1.58 (m, 1H), 1.87–1.88 (br d, J = 14.09 Hz, 2H), 1.97–2.07 (m, 1H) 2.67–2.76 (m, 4H), 3.27 (m, 1H), 5.27 (s, 2H), 5.90 (s, 2H), 6.44–6.46 (dd, J = 2.54, 8.41, 1H), 6.61 (d, J = 2.74 Hz, 1H), 6.70–6.72 (d, J = 8.22 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ ppm 29.5, 30.9, 31.9, 43.1, 54.2, 61.7, 98.2, 101.4, 106.3, 107.5, 142.9, 148.5, 153.1, 168.7, 175.7; IR (thin film): 2918, 2697, 2359, 1487, 1184 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C17H22N3O4 332.1605, found 332.1597; HPLC purity >95%.
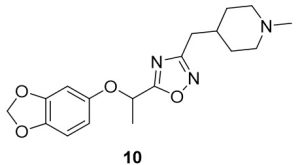
5-(1-(Benzo[d][1,3]dioxol-5-yloxy)ethyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (10). Compound 2 (107.4 mg, 0.51 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (88.3 mg, 0.52 mmol) were reacted according to General Procedure B to furnish the oxadiazole 10 as a yellow solid (64.8 mg, 0.19 mmol, 37% yield). Rf = 0.40 (10% MeOH in DCM); mp = 80–82 ◦C; 1H NMR (400 MHz, methanol-d4) δ ppm 1.32 (m, 2H), 1.68 (br d, J = 11.74 Hz, 2H), 1.73 (d, J = 6.65 Hz, 3H), 1.75–1.86 (m, 1H) 2.07 (br t, J = 11.74 Hz, 2H), 2.30 (s, 3H), 2.68 (d, J = 7.04 Hz, 2H), 2.90 (br d, J = 12.13 Hz, 2H), 5.56 (q, J = 6.65 Hz, 1H), 5.90 (s, 2H), 6.42 (dd, J = 8.61, 2.35 Hz, 1H), 6.58 (d, J = 2.35 Hz, 1H), 6.68 (d, J = 8.61 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ 19.8, 32.4, 46.3, 56.3, 71.0, 100.8, 102.7, 108.8, 109.5, 144.4, 149.7, 153.7, 170.3, 180.1; IR (thin film): 2932, 2779, 2359, 1483, 1180 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C18H24N3O4 346.1761, found 346.1757; HPLC purity >95%.
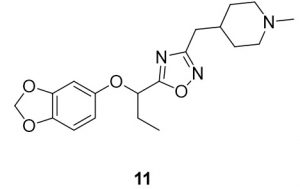
5-(1-(benzo[d][1,3]dioxol-5-yloxy)propyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (11). Compound 3 (114.3 mg, 0.51 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (88.9 mg, 0.52 mmol) were reacted according to General Procedure B to furnish the oxadiazole 11 as a pale yellow solid (87.3 mg, 0.24 mmol, 48% yield). Rf = 0.40 (10% MeOH in DCM); mp = 61–63 ◦C; 1H NMR (400 MHz, methanol-d4) δ ppm 1.05 (t, J = 7.43 Hz, 3H) 1.33 (qd, J = 12.39, 3.91 Hz, 2H) 1.64 (br dd, J = 13.11, 2.15 Hz, 2H) 1.71–1.84 (m, 1H) 2.01 (br t, J = 11.93 Hz, 2H) 2.05–2.18 (m, 2H) 2.26 (s, 4H) 2.67 (d, J = 7.04 Hz, 2H) 2.85 (br d, J = 11.74 Hz, 2H) 5.36 (t, J = 6.65 Hz, 1H) 5.89 (s, 2H) 6.39 (dd, J = 8.41, 2.54 Hz, 1H) 6.56 (d, J = 2.74 Hz, 1H) 6.66 (d, J = 8.61 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ ppm 8.2, 26.9, 31.0, 31.5, 33.3, 44.8, 54.9, 74.7, 99.2, 101.3, 107.5, 107.9, 143.0, 148.4, 152.7, 168.9, 178.2; IR (thin film): 2917, 2784, 2359, 1485, 1182 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C19H26N3O4 360.1918, found 360.1912; HPLC purity >95%.
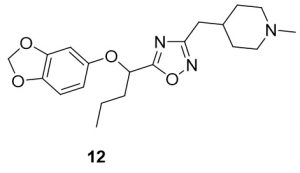
5-(1-(benzo[d][1,3]dioxol-5-yloxy)butyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (12). Compound 4 (107.4 mg, 0.45 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (84.9 mg, 0.49 mmol) were reacted according to General Procedure B to furnish the oxadiazole 12 as a pale yellow solid (36.9 mg, 0.10 mmol, 22% yield). Rf = 0.44 (10% MeOH in DCM); mp = 66–68 ◦C; 1H NMR (400 MHz, methanol-d4) δ ppm 0.97–1.00 (t, J = 7.43 Hz, 3H) 1.27–1.37 (m, 2H) 1.41–1.57 (m, 2H), 1.61–1.65 (m, 2H), 1.71–1.82 (m, 1H), 1.94–2.03 (m, 3H), 2.06–2.15 (m, 1H), 2.25 (s, 3H), 2.65–2.66 (d, J = 7.04 Hz, 2H), 2.83–2.85 (br d, J = 11.74 Hz, 2H), 5.39 – 5.42 (dd, J = 5.87, 7.43 Hz, 1H), 5.89 (s, 2H), 6.36 – 6.39 (dd, J = 2.35, 8.61 Hz, 2H), 6.54–6.55 (d, J = 2.74 Hz, 1H), 6.64–6.66 (d, J = 8.61 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ ppm 12.6, 17.9, 31.0, 31.5, 33.4, 35.7, 44.9, 54.9, 73.3, 99.2, 101.3, 107.5, 107.8, 143.0, 148.4, 152.7, 168.9, 178.4; IR (thin film): 2917, 2784, 2359, 1485, 1182 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C20H28N3O4 374.2074, found 374.1013; HPLC purity >99%.
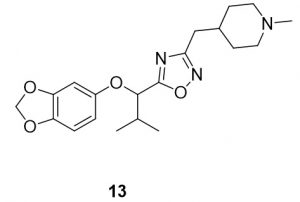
5-(1-(benzo[d][1,3]dioxol-5-yloxy)-2-methylpropyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (13). Compound 5 (93.6 mg, 0.39 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (75.6 mg, 0.44 mmol) were reacted according to General Procedure B to furnish the oxadiazole 13 as a pale yellow solid (59.7 mg, 0.39 mmol, 41% yield). Rf = 0.44 (10% MeOH in DCM); mp = 58–60 ◦C; 1H NMR (400 MHz, methanol-d4) δ ppm 0.98 (d, J = 7.04 Hz, 3H), 1.13 (d, J = 6.65 Hz, 3H), 1.24–1.43 (m, 3H), 1.57–1.68 (m, 2H), 1.71–1.85 (m, 1H), 2.00 (tt, J = 11.93, 2.74 Hz, 2H), 2.23–2.29 (m, 4H), 2.38 (sxt, J = 1.00 Hz, 1H), 2.67 (d, J = 7.43 Hz, 2H), 2.85 (br d, J = 11.74 Hz, 2H), 5.14 (d, J = 6.65 Hz, 1H), 5.89 (s, 2H),6.32–6.40 (m, 1H), 6.54 (d, J = 2.35 Hz, 1H), 6.65 (d, J = 8.61 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ ppm 16.9, 17.0, 31.0, 31.5, 32.7, 33.4, 44.9, 54.9, 78.7, 99.1, 101.3, 107.5, 107.7, 142.9, 148.4, 152.9, 168.9, 177.8; IR (thin film): 2931, 2359, 1484, 1181, 1035 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C20H28N3O4 374.2074, found 374.2066; HPLC purity >96%.
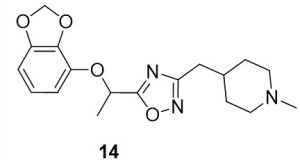
5-(1-(benzo[d][1,3]dioxol-4-yloxy)ethyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (14). Compound 6 (38.6 mg, 0.18 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (34.9 mg, 0.20 mmol) were reacted according to General Procedure B to furnish the oxadiazole 14 as a pale yellow solid (9.9 mg, 0.03 mmol, 16% yield). Rf = 0.25 (10% MeOH in DCM); mp = 78–80 ◦C; 1H NMR (400 MHz, methanol-d4) δ ppm 1.33 (qd, J = 3.5, 12.4 Hz, 2H), 1.68 (br d, J = 11.74 Hz, 2H), 1.73 (d, J = 6.65 Hz, 3H), 1.75–1.86 (m, 1H) 2.07 (br t, J = 11.74 Hz, 2H), 2.30 (s, 3H), 2.68 (d, J = 7.04 Hz, 2H), 2.90 (br d, J = 12.13 Hz, 2H), 5.56 (q, J = 6.65 Hz, 1H), 5.90 (s, 2H), 6.42 (dd, J = 8.61, 2.35 Hz, 1H), 6.58 (d, J = 2.35 Hz, 1H), 6.68 (d, J = 8.61 Hz, 1H); 13C NMR (126 MHz, methanol-d4) δ ppm 19.8, 32.4, 46.3, 56.3, 71.0, 100.8, 102.7, 108.8, 109.5, 144.4, 149.7, 153.7, 170.3, 180.1; IR (thin film): 2936, 2779, 2359, 1457, 1283 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C18H24N3O4 346.1761, found 346.1756; HPLC purity >95%.
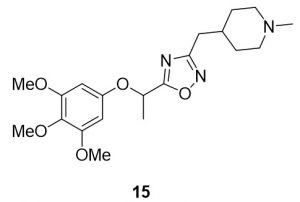
3-((1-methylpiperidin-4-yl)methyl)-5-(1-(3,4,5-trimethoxyphenoxy)ethyl)-1,2,4-oxadiazole (15). Compound 7 (128.7 mg, 0.50 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (88.8 mg, 0.52 mmol) were reacted according to General Procedure B to furnish the oxadiazole 15 as a red oil (109.8 mg, 0.28 mmol, 56% yield). Rf = 0.25 (10% MeOH in DCM); 1H NMR (400 MHz, methanol-d4) δ ppm 1.31–1.40 (m, 2H), 1.65–1.68 (m, 2H), 1.73–1.82 (m, 5H), 2.00–2.05 (br t, J = 11.1 Hz, 2H), 2.28 (s, 2H), 2.68–2.70 (d, J = 7.0 Hz, 2H), 2.86–2.89 (d, J = 11.9 Hz, 2H), 3.70 (s, 3H), 3.80 (s, 6H), 5.71– 5.75 (q, J = 6.61 Hz, 1H), 6.33 (s, 2H); 13C NMR (126 MHz, methanol-d4) δ ppm 18.4, 31.0, 31.5, 33.4, 44.8, 54.9, 55.2, 59.8, 68.6, 93.9, 132.9, 153.6, 153.8, 169.0, 178.7; IR (thin film): 2933, 2359, 2341, 1504, 1126 cm-1; HRMS (ESI) m/z: [M+H]+ Calcd for C20H30N3O5 392.2180, found 392.2171; HPLC purity >95%.
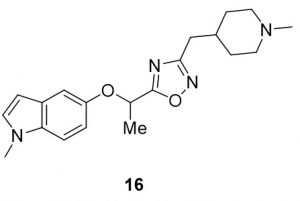
5-(1-((1-methyl-1H-indol-5-yl)oxy)ethyl)-3-((1-methylpiperidin-4-yl)methyl)-1,2,4-oxadiazole (16). Compound 8 (46.3 mg, 0.21 mmol) and (Z)-N’-hydroxy-2-(1-methylpiperidin-4-yl)acetimidamide (39.1 mg, 0.23 mmol) were reacted according to General Procedure B to furnish the oxadiazole 16 as a yellow oil (31.5 mg, 0.09 mmol, 42%). Rf = 0.19 (10% MeOH in DCM); 1H NMR (400 MHz, methanol-d4) δ ppm 1.19-1.32 (m, , 2H), 1.54-1.56 (br d, J = 7.63 Hz, 2H), 1.65-1.75 (m, 4H), 1.86-1.92 (td, J = 1.96, 11.93 Hz, 2H), 2.21 (s, 3H), 2.60-2.62 (d, J = 7.04 Hz, 2H), 2.74-2.77 (br d, J = 11.74 Hz, 2H), 3.73 (s, 3H), 5.58-5.62 (q, J = 6.39 Hz, 1H), 6.28-6.29 (d, J = 3.13 Hz, 1H), 6.84-6.87 (dd, J = 2.54, 8.80 Hz, 1H), 7.07-7.10 (dd, J = 3.13, 7.83 Hz, 1H), 7.21-7.24 (d, J = 9.00, 1H); 13C NMR (126 MHz, methanol-d4) δ 18.6, 31.0, 31.6, 33.4, 44.9, 54.9, 69.9, 100.0, 106.6, 109.5, 112.8, 128.8, 129.7, 133.3, 151.0, 168.9, 179.3; IR: 2932, 2779, 2359, 2341, 1482, 1179; HRMS Calcd. For C20H27N4O2 [M + H]+ 355.2129, found 355.2121; HPLC purity >98%.
ACKNOWLEDGEMENTS
This work was supported by the National Institutes on Aging TREAT-AD Center for Alzheimer’s Disease grant U54 AG065187.
REFERENCES
- Katis, V.; Bradshaw, W.; Betarbet, R.; Fu, H.; DU, Y.; Qian, K.; Gileadi, O. MSN (Moesin): A Target Enabling Package https://zenodo.org/record/4429232 (accessed Jan 27, 2022).
- Biernacki, K.; Dasko, M.; Ciupak, O.; Kubinski, K.; Rachon, J.; Demkowicz, S. Novel 1,2,4-Oxadiazole Derivatives in Drug Discovery. Pharmaceuticals 2020, 13, 111.
- (a) Diana, G.; Volkots, D.; Nitz, T.; Bailey, T.; Long, M.; Vescio, N.; Aldous, S.; Pevear, D.; Dutko, F. Oxadiazoles as Ester Bioisosteric Replacements in Compounds Related to Disoxaril. Antirhinovirus Activity. Journal of Medicinal Chemistry1994, 37, 2421-2436. (b) Omar, F.; Mahfouz, N.; Rahman, M. Design, Synthesis and Antiinflammatory Activity of Some 1,3,4-Oxadiazole Derivatives. European Journal of Medicinal Chemistry 1996, 31, 819-825.
- Sui, G.; Song, X.; Zhang, B.; Wang, Y.; Liu, R.; Guo, H.; Wang, J.; Chen, Q.; Yang, X.; Hao, H. et al. Design, Synthesis And Biological Evaluation Of Novel Neuchromenin Analogues As Potential Antifungal Agents. European Journal of Medicinal Chemistry2019, 173, 228-239.
- Katritzky, A.; Shestopalov, A.; Suzuki, K. A Convenient Synthesis Of Chiral 1,2,4-Oxadiazoles From N-Protected (Α-Aminoacyl)Benzotriazoles. Arkivoc2005, 2005, 36-55.
- Porcheddu, A.; Cadoni, R.; De Luca, L. A Fast and Efficient One-Pot Microwave Assisted Synthesis of Variously Di-Substituted 1,2,4-Oxadiazoles. Organic & Biomolecular Chemistry2011, 9, 7539.