In my last post, I told you I had soaked my follow-up compounds into HAO1 crystals to start developing structural explanations of how these compounds work. So far I have analysed four interesting protein-compound structures: 2 at the active site, 1 at the gating loop and 1 at the oligomeric interface. Full experimental details can be found in my Zenodo post.
The first structure had a compound derived from fragment 1 bound at the active site. This compound (5-bromo-N-methyl-1H-indazole-3-carboxamide; FUP1A for short) expands fragment 1 in two directions: adding a bulky, hydrophobic bromine to the aromatic ring and adding a methyl (CH3) to the carboxamide (CO-NH) at the other end of the molecule (figure 1C). FUP1A shows more typical displacement of the substrate-coordinating tryptophan (Trp110), rotating this residues by 180° so that it points away from the substrate binding pocket (figure 1A). FUP1A binds further into the substrate binding pocket than the fragment, likely due to the addition of the bulky bromine, so that it is now in the same orientation as the published HAO1 inhibitors (CDST and CCPST, PDB codes 2RDT and 6GMC respectively) as illustrated in figure 1D.
When taken together with the potent activity assay result – an IC50 of 62 µM, 7X improvement over fragment 1 – for a similar compound (6-bromo-N-ethyl-1H-indazole-3-carboxamide; FUP1B; structure shown in figure 1D) with the bromine one position away and a longer extension (ethyl, CH2CH3, rather than methyl) on the other side, this structure opens up some interesting questions. Does the orientation of fragment 1 reflect a more optimal position for the core scaffold? If so, does the ethyl group and/or the different position of the ethyl group of FUP1B allow it to maintain this position? Or do one or both of these groups disrupt the active site residues in an entirely different way? To answer these questions, I am currently working to obtain a structure with FUP1B bound.
The second structure was with a compound derived from fragment 2, also at the active site. This compound (6-amino-1-benzyl-5-(methylamino) pyrimidine-2, 4(1H, 3H)-dione; FUP2A) was chosen to explore alternate groups for the second half of fragment 2, replacing the original ring (cyclohexane) with an aromatic one (benzene), since the former did not seem to contribute to fragment binding. FUP2A did not show much improvement in the activity assay over fragment 2 and this is reflected by the bound structure, which shows that FUP2A and fragment 2 bind in very similar poses with the same displacement of Trp110 (figure 1B). As well as validating this scaffold as a consistent HAO1-binder, this follow up structure suggests that the best way to continue development would be to expand from other positions on the substituted ring, to better disrupt active site residues.
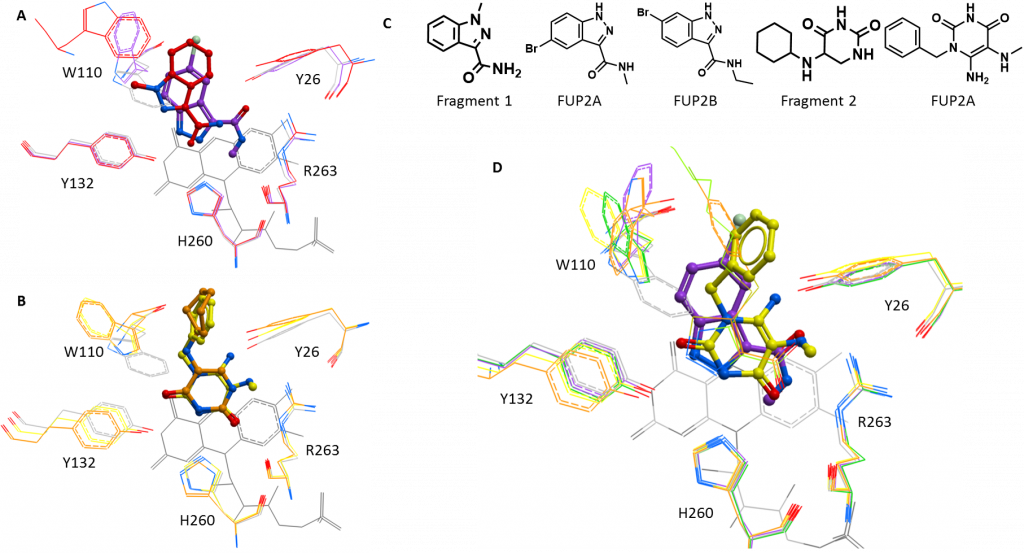
Figure 1: New structures of active site inhibitors of HAO1. A. Fragment 1 (red) and the related compound, FUP1A (purple). B. Fragment 2 (orange) and the related compound, FUP2A (yellow). C. Chemical structures of interesting compounds. D. Comparison of FUP1A (purple) and FUP2A (yellow) with the published inhibitors, CDST (green) and CCPST (orange), shown as sticks.
The third structure is of a compound bound at the gating loop and derived from fragment 5 (figure 2B). This compound (2-((4H-1, 2, 4-triazol-3-yl) thio)-1-(4-(3-chlorophenyl) piperazin-1-yl) ethan-1-one; FUP5A) replaces the fluoride on the original phenyl ring with a chloride at a different position (4-fluoro to 3-chloro) and extends the opposite end of the compound with a sulphur and a nitrogen-rich aromatic ring (triazole) (Figure 3B). FUP5A superimposes fairly well with its parent fragment at the piperazine (six-membered, non-aromatic ring with a nitrogen at either end) with the piperazine displacing the sidechain of Tyr134 and the substituted phenyl ring continuing to disrupt the gating loop (Figure 2A). the extension at the opposite end of the molecule does not cause the same conformational change (shift of helix 4) but rather points mostly out towards the solvent, not forming further interactions with the protein (Figure 3A). This structure suggests that larger extensions from the phenyl end may be more effective in disrupting the gating loop but care is needed in extending the other end of the molecule to avoid losing efficiency by including non-interacting chemical groups.
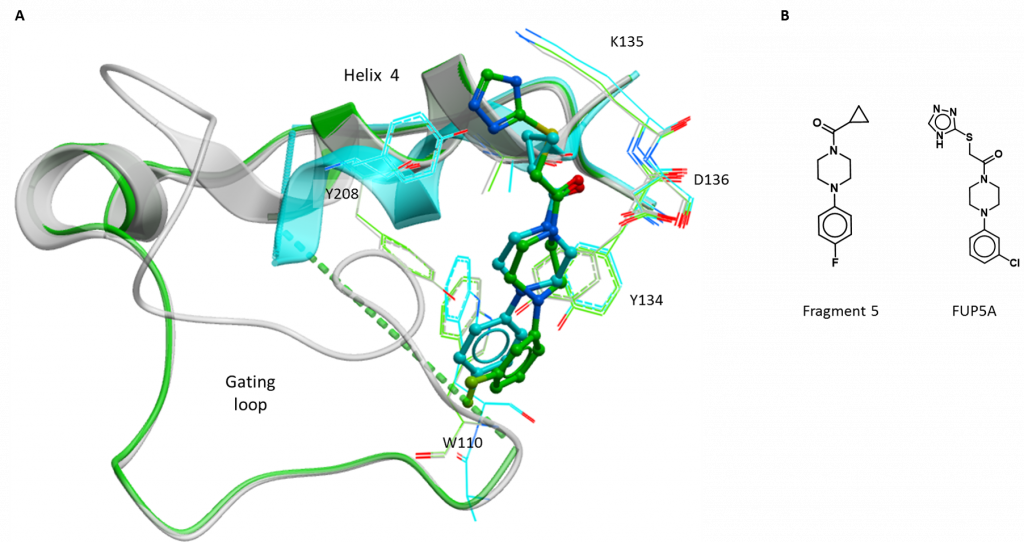
Figure 2: New structure of gating loop site inhibitor of HAO1. A. Fragment 5 (blue) and the related compound, FUP5A (green). B. Chemical structures of interesting compounds.
The final structure has a compound derived from fragment 6 bound at the subunit interface. This compound (1-(5-chloro-pyrimidin-4-yl)-4-(pyridin-2-yl)-piperazine; FUP6A) extends the fragment scaffold, essentially in a straight line, by the addition of two further rings and the structure shows that it binds in the same manner – through hydrophobic interactions with both subunits and pi-stacking with the ring of Tyr163 on one subunit (figure 3A). The fact that FUP6A can bind with the HAO1 tetramer intact explains why it did not show much inhibition in the activity assay and this structure also helps to further map the binding pocket at the interface. Taken together with the more potent follow up compounds (e.g. FUP6B, FUP6C, shown in figure 3B), this structure validates the need for bulky additions perpendicular to the core scaffold (like the two fluorides in FUP6B) to more effectively disrupt the HAO1 tetramer.
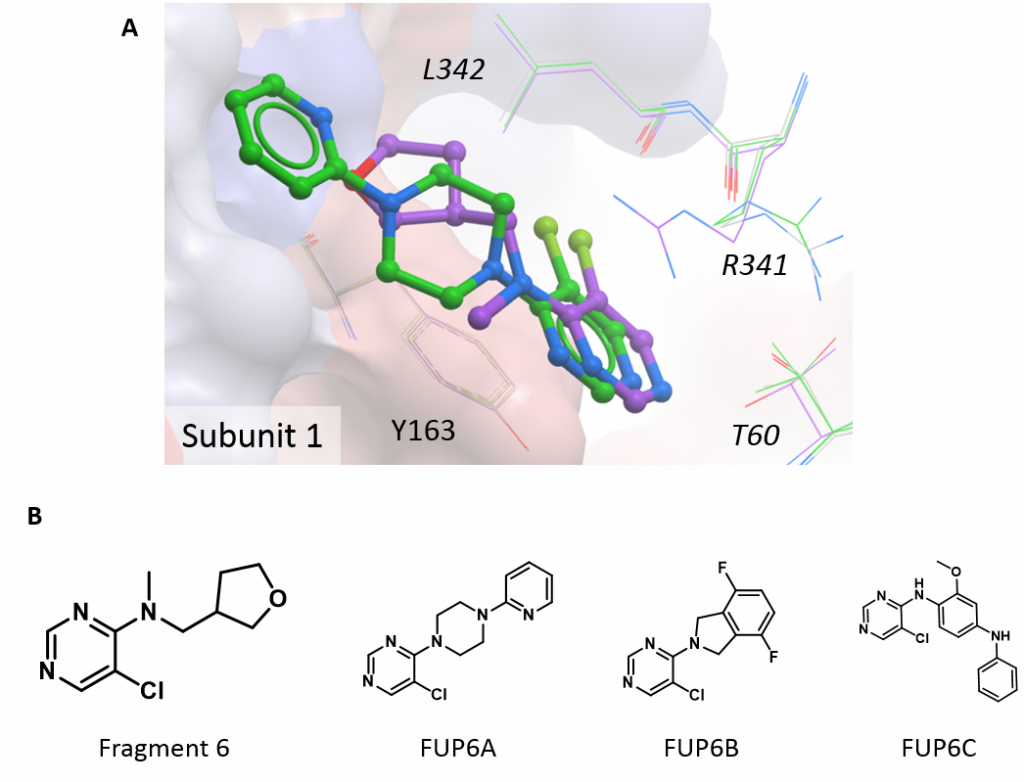
Figure 3: New structure of interface site inhibitor of HAO1. A. Fragment 6 (purple) and the related compound, FUP6A (green). B. Chemical structures of interesting compounds.
Since some compounds may require greater movements of the gating loop or tetramer subunits than can be freely accommodated within preformed crystals, I am also trying to co-crystallise with the most promising hits. This involves adding one compound per aliquot of freshly purified HAO1 protein before setting up trials to screen a wide range of crystallisation conditions. Co-crystallisation experiments requiring more time and resources than soaking but do have a few benefits: a greater number of conditions can be tested to look for those that only yield crystals with a given ligand; a lower concentration of compound can be used to achieve an equivalent saturation of the protein; and binding of the compound occurs in solution, accommodating a greater degree of flexibility. To this end, I have set up crystal plates with the 13 top compounds (4 active site, 6 gating loop and 3 interface), which have yielded a number of likely-looking crystals that I will be screening over the coming weeks.