Histone decaetylases (HDACs) are one of the several enzymes that take part in regulating gene expression. They catalyze the removal of acetyl groups from ε-N-acetylated lysines from both, histone and non-histone protein substrates. By doing this, they maintain a balance with respect to the activity of the acetyltransferases in the cells. The HDAC family consists of 18 members, which is divided into 4 classes: Class I (HDAC1, 2, 3, 8), Class IIa (4, 5, 7, 9), Class IIb (6, 10), Class III/sirtuins (Sirt1-7) and Class IV (HDAC11). Class I, II and IV enzymes are zinc-dependent, while that of Class III are NAD+-dependent (Table 1).
Table 1. Classification of HDACs (Zhang et al., 2017).
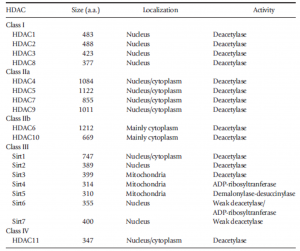
HDACs have been reported to be dysregulated in several cancers (Bassett and Barnett, 2014; Li and Seto, 2016; Zhang et al., 2017) and have been associated with other non-cancerous diseases and disorders (Roche and Bertrand, 2016) (Table 2).
Table 2. Association of HDACs with some cancers (Zhang et al., 2017).

HDAC inhibitors:
Due to their relevance and involvement in a diverse range of diseases and disorders, HDACs are important drug targets. Several attempts have been and are being made across the globe to develop HDAC inhibitors (HDACi) that would possess high potency and least toxicity. So far, only five HDACi have been approved for clinical application- SAHA (cutaneous T-cell lymphoma), Belinostat (peripheral T-cell lymphoma), Panabiostat (multiple myeloma), Romidepsin (cutaneous T-cell lymphoma) and Valproic acid (epilepsia, biopolar disorder and migraine). All the five approved drugs are either pan inhibitors (HDAC specificity against Class I, II and IV, in case of SAHA, Belinostat and Panabiostat) or target a specific class of HDAC (Class I in case of Romidepsin and Class I, IIa in case of Valproic acid), thus, lacking specificity against a specific type of HDAC. In addition, several molecules are under different stages of preclinical or clinical trials, for various cancers and other disorders such as brain disorders and aging (Eckschlager et al., 2017; Ganai et al., 2016; Pasyukova and Vaiserman, 2017; Qiu et al., 2017). Several studies have also reported the development of selective drugs, but their extensive preclinical and clinical evaluation is underway (Qin et al., 2017; Thaler and Mercurio, 2014).
Challenges in the development of HDAC inhibitors:
There are two main challenges that need to be overcome in developing drug molecules against HDACs.
- Selectivity: As an expected and obvious approach, the active sites of HDACs have been the first to be targeted. But since, the overall structure of HDAC active sites is similar, this has led to HDACi that are not specific for an HDAC in their inhibition. Since, these inhibitors target all (or most of) HDACs, they increase the risk of toxicity and thus, side-effects of the drugs. Some of the side effects include gastrointestinal disturbances, fatigue and QT prolongation with risk of fatal arrhythmia. Having less toxic, selective drug molecules would be a great advancement for therapeutics. Here, being able to target the subtle differences in the areas of the active site or the adjoining areas following designing of compounds using structure-guided approaches is an upcoming approach (Di Giorgio et al., 2015; Maolanon et al., 2016).
Also, since HDACs interact with several protein partners and are a part of several multi-protein complexes, novel strategies intend to target the binding interfaces of these interactions and disrupting the protein complexes to render the enzyme non-functional (inactive) when devoid of its partners. Differences in these interactions and interaction partners could bring selectivity in the targeting of most HDACs if not all (Di Giorgio et al., 2015; Maolanon et al., 2016).
- Assay development: Appropriate high-throughput assays for monitoring HDAC activity (activity-based assays) or their interactions with their binding partners (binding-based assays) serve as the platforms that are exploited for the screening of massive compound libraries. Thus, appropriately designing and optimizing assays with best possible read-out windows and reproducibility is of utmost importance in the screening process.
Despite having an overall similar active site, HDACs show variations in catalyzing the canonical substrates. Moreover, some HDACs are less active than the others. Due to these variations, being able to identify the apt substrate for the HDAC in hand and being able to use it in a format to get a reasonable quantifiable activity to develop an assay is a crucial step. Few studies performed using a substrate panel to monitor the activity of different HDACs serve as a good starting point for the activity-based assays (Madsen and Olsen, 2012).
Similarly, identifying the best interaction partner and being able to develop an appropriate assay with it is also important.
I focus my work on the development of in vitro high-throughput biophysical assays that could serve as a platform for the screening of small-molecule compound libraries. To start off, I will work with HDAC11 and HDAC4 at the moment.
References:
- Bassett, S.A., and Barnett, M.P. (2014). The role of dietary histone deacetylases (HDACs) inhibitors in health and disease. Nutrients 6, 4273-4301.
- Di Giorgio, E., Gagliostro, E., and Brancolini, C. (2015). Selective class IIa HDAC inhibitors: myth or reality. Cellular and molecular life sciences : CMLS 72, 73-86.
- Eckschlager, T., Plch, J., Stiborova, M., and Hrabeta, J. (2017). Histone Deacetylase Inhibitors as Anticancer Drugs. International journal of molecular sciences 18.
- Ganai, S.A., Ramadoss, M., and Mahadevan, V. (2016). Histone Deacetylase (HDAC) Inhibitors – emerging roles in neuronal memory, learning, synaptic plasticity and neural regeneration. Curr Neuropharmacol 14, 55-71.
- Li, Y., and Seto, E. (2016). HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harbor perspectives in medicine 6.
- Madsen, A.S., and Olsen, C.A. (2012). Profiling of substrates for zinc-dependent lysine deacylase enzymes: HDAC3 exhibits decrotonylase activity in vitro. Angewandte Chemie 51, 9083-9087.
- Maolanon, A.R., Madsen, A.S., and Olsen, C.A. (2016). Innovative Strategies for Selective Inhibition of Histone Deacetylases. Cell chemical biology 23, 759-768.
- Pasyukova, E.G., and Vaiserman, A.M. (2017). HDAC inhibitors: A new promising drug class in anti-aging research. Mechanisms of ageing and development 166, 6-15.
- Qin, H.T., Li, H.Q., and Liu, F. (2017). Selective histone deacetylase small molecule inhibitors: recent progress and perspectives. Expert opinion on therapeutic patents 27, 621-636.
- Qiu, X., Xiao, X., Li, N., and Li, Y. (2017). Histone deacetylases inhibitors (HDACis) as novel therapeutic application in various clinical diseases. Progress in neuro-psychopharmacology & biological psychiatry 72, 60-72.
- Roche, J., and Bertrand, P. (2016). Inside HDACs with more selective HDAC inhibitors. European journal of medicinal chemistry 121, 451-483.
- Thaler, F., and Mercurio, C. (2014). Towards selective inhibition of histone deacetylase isoforms: what has been achieved, where we are and what will be next. ChemMedChem 9, 523-526.
- Zhang, H., Shang, Y.P., Chen, H.Y., and Li, J. (2017). Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatology research : the official journal of the Japan Society of Hepatology 47, 149-159.