Introduction
In this blog post I will share details on the new molecules we have designed and synthesized or purchased based on one of our original hits from the x-ray crystallography fragment screen. The compound is called F9000505 and it is bound to pocket B. The crystal structure was deposited in the Protein Data Bank (pdb) with PDB ID 5QRU. This post will give you a feel for some of the ways we are approaching fragment optimization. Posts in the near future will describe strategies at other fragment sites and also provide data on the binding of these compounds to brachyury as it emerges.
Background / Project Goal
The expression of brachyury (TBXT) is a hallmark of chordoma. A growing body of data suggests that inhibition of TBXT protein activity, expression, or interactions with protein partners are predicted to be effective and specific means for treating chordoma. A key aim of this project is to discover compounds that bind directly to brachyury with sufficient affinity so that they ca be optimized into clinical candidates for the treatment of chordoma.
Initial Fragment Optimization Strategy
Fragment-based screening by x-ray crystallography is very powerful in that the location of the fragment hits on the target protein (brachyury) are known after the experiment, and the structure provides details of the interactions that the fragments make with brachyury. Crystallography can identify fragments that bind very weakly, so there is often a long way to go to grow the hit molecules into compound that bind potently enough to elicit the desired biological effects. We are taking an aggressive approach to exploring each of our hits so that we can soon focus efforts on the most promising molecules and binding sites.
The two sources of new molecules are purchase of commercially available compounds, and design and synthesis of new compounds. Purchase of compounds is cost effective and rapid in the early stages of a project but becomes limited as molecules become more complicated to capture the intricacies of the binding sites.
We have purchased compounds primarily through a company called ChemSpace. We performed substructure and similarity searches in the ChemSpace available compound database for each x-ray crystallography hit (“query” molecules for the searching). A substructure search means specific details of the hit need to be present in the molecules identified by the search. A similarity search relaxes the criteria and allows for selection of molecules that the computer tells us have certain similarities to our query molecules. These similarities could be in categories like size, shape, and electronic properties. Similarity searches can identify compounds that are more different from the hits which may be important if they identify new interactions, scaffolds that are easier to make new analogs of, or compound that explore different vectors (directions) around the hits.
We chose initial analogues for purchase to provide important information around each fragment in several domains:
- Compounds with functional groups that will serve as efficient handles for further elaboration.
- Compounds that explore growth of the hit fragment in logical directions based on the crystal structure.
- Compounds that add additional functionality that can make new interactions in order to enhance potency.
- Compounds that teach us where linkers may be added in order to develop brachyury degraders.
By making the fragments larger we can understand where growth is beneficial to potency. This will provide focus to future rounds of purchase or chemistry. Incorporation of chemical handles that can be rapidly elaborated with a variety of building blocks will allow us to make additional analogs quickly. Adding functionality (for example amides, ureas, phenols, heterocycles, substituted phenyls) that can make energetically favorable interactions (hydrogen bonds, pi-pi stacking, lipophilic interactions, charge-charge interactions) improves our chances of increasing potency. Utilizing commercially available analogs of the initial fragments found bound to the TBXT crystal structure provides the means to quickly generate structure activity relationships (SAR – this means how changing the structure changes how well the compound binds to brachyury) around each fragment. Initial SAR is critical as it provides confidence around each hit (can it be optimized, do several similar compounds also bind) and aids in site selection to narrow our focus.
Fragment F9000505, binding site B, PDB ID: 5QRU
Compounds for purchase
Figure 1 depicts all the compounds that we could buy identified from substructure and similarity searching around fragment hit F9000505. This compound binds to site B, and more details on the way it binds will be found below in the new compound design section. Each dot represents a compound and they are organized in the plot according to two easy to calculate and important properties: lipophilicity (clogP) and molecular weight (MW). The green circles are the compounds that we have purchased and are screening. If any of these are active, we can purchase other compounds (blue dots) that can further supplement the SAR. This iterative cycle of design, synthesis (purchase if possible), and test is the life blood of medicinal chemistry compound optimization.
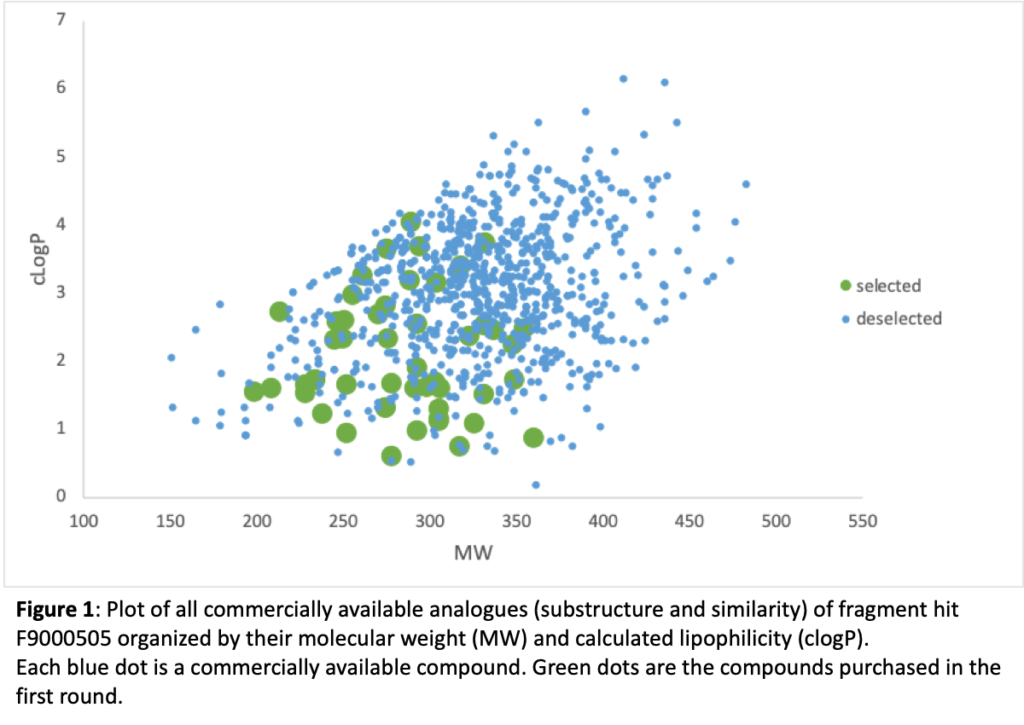
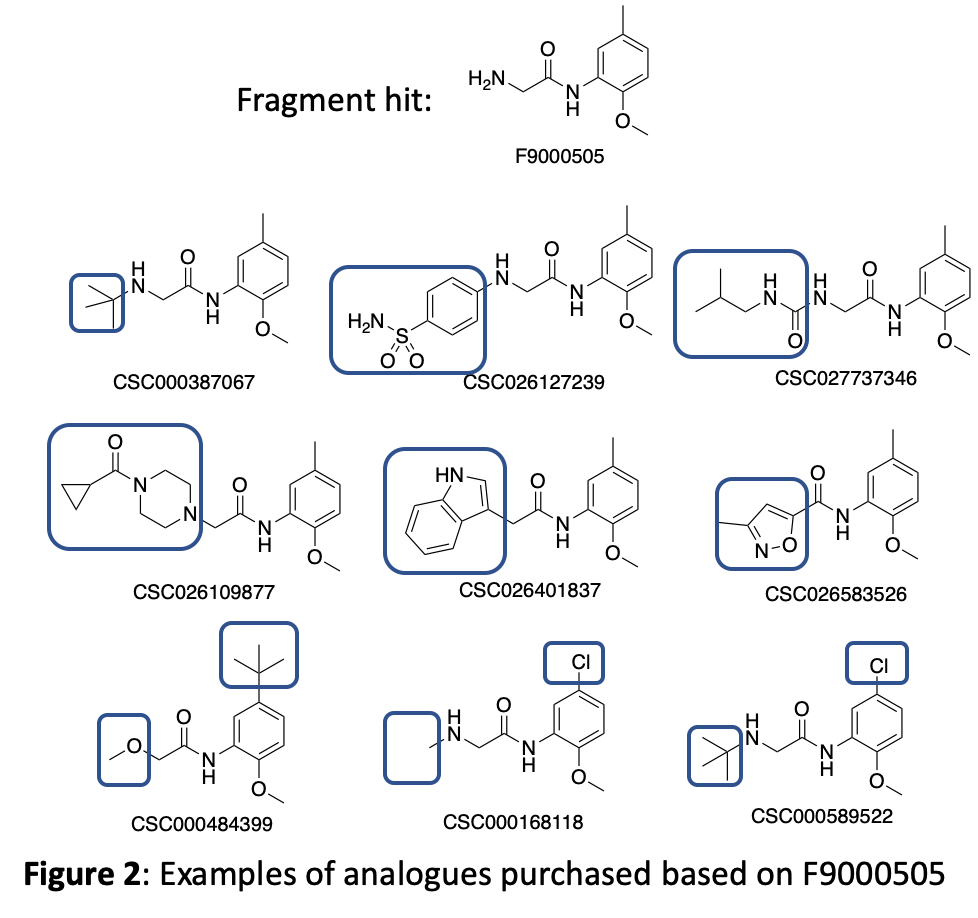
Figure 2 depicts examples of molecules we purchased from this set that demonstrate the availability of useful molecules that can answer some of the specific medicinal chemistry questions we need to ask. The blue boxes highlight the areas with structural modifications to the hit compound.
Design and Synthesis of New Compounds
We have chemists on our team at UNC and also a talented team of chemists at a company called Piramal. We have been able to make excellent progress on new compound synthesis even with the initial lab shutdowns and then the reduced capacity necessitated by Covid-19. The team at Piramal has focused on preparation of analogs of fragments for which there are fewer commercial analogs, and on more elaborate chemistry to explore promising fragment sites in greater detail using the same principles outlined above.
Figure 3 depicts the crystal structure of fragment F9000505 bound to pocket B of brachyury. This simple compound results from the acylation of 2-methoxy-5-methylaniline with the amino acid glycine. The phenyl ring makes lipophilic contacts with the protein and the 5-methyl group tucks into a lipophilic pocket. The oxygen of the 2-methoxy group makes an intramolecular hydrogen bond with the adjacent NH, helping to orient the trajectory of the carbonyl of the amide and also the terminal amine of the glycine. In this locked in orientation the carbonyl of the fragment forms a hydrogen bond with Ala119 of brachyury and the primary amine forms a hydrogen bond to the carbonyl of Pro117.
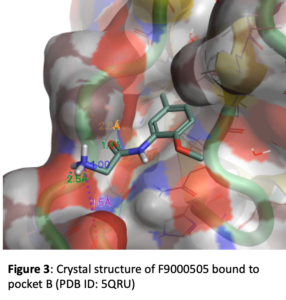
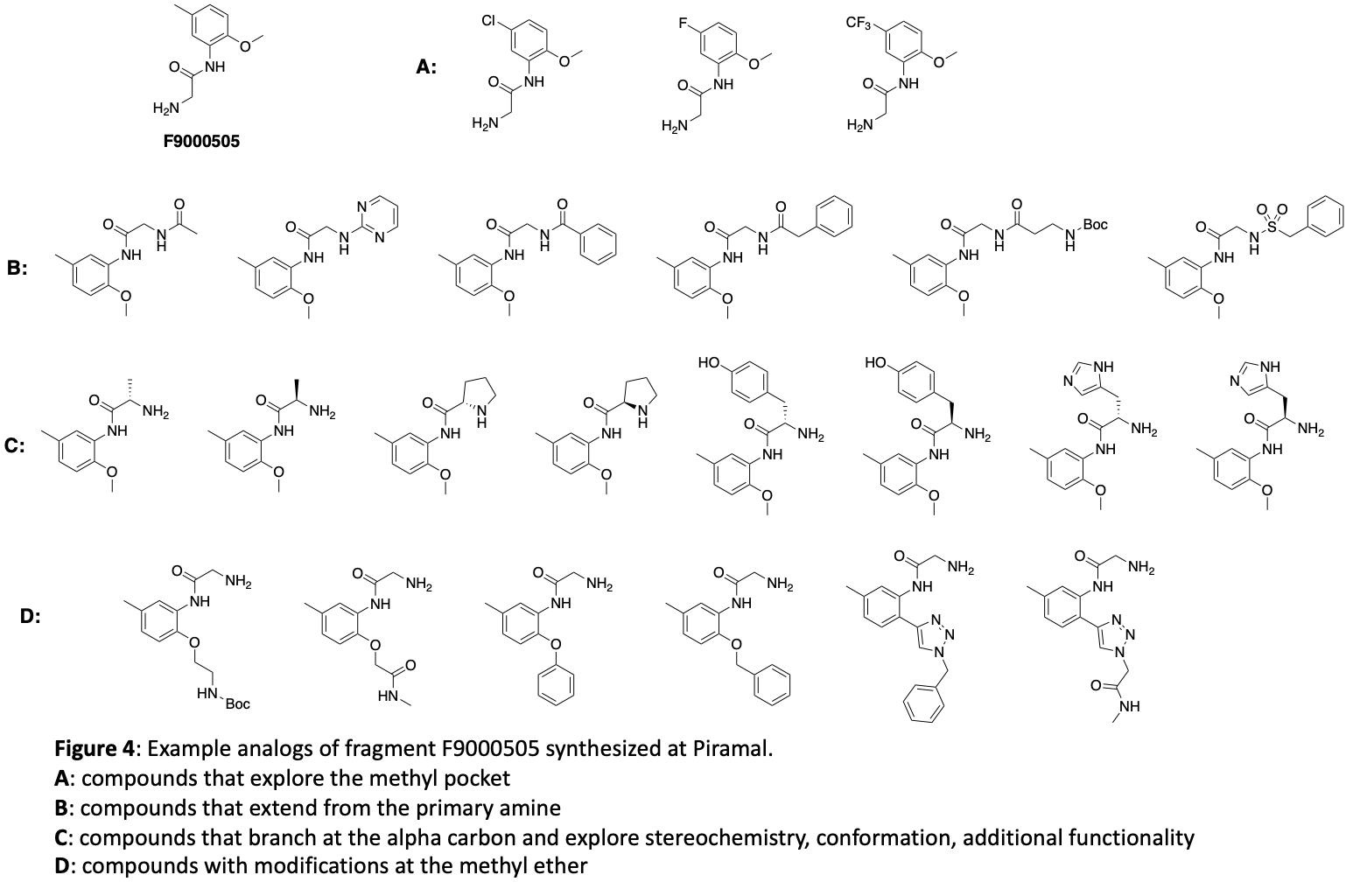
Based on this structure there are a number of avenues that need to be explored for optimization of binding. The methyl substituent in the lipophilic pocket may not be ideal. Groups with different size and electronic characteristics need to be explored (for example: F, Cl, ethyl, cyclopropyl). There is plenty of room to extend off of the methoxy group. Doing this may allow us to pick up additional interactions with the surface of the protein. In addition, this may be an attractive position for a linker that will be suitable for synthesis of a bifunctional degrader. Another strategy is to extend the primary amine in order to reach Glu116 which could increase affinity. The alpha carbon of the glycine moiety presents another opportunity. Simple substituents like methyl will add conformational constraint which can enhance potency. Because this building block comes from amino acids, there is opportunity to quickly explore stereochemistry at this position and a range of functional groups that have potential to form new interactions. Finally, we can form a 5- or 6-membered ring between the NH and the O of the fragment. This will serve to further lockdown the orientation of the carbonyl and NH2 that make key hydrogen bonds and offer different vectors for exploring interactions with other brachyury residues. Our initial compound design targeted 68 molecules. These molecules have all been synthesized and submitted from screening in the surface plasmon resonance (SPR) binding assay. A sampling of the 68 compounds are depicted in Figure 4. These show example modifications of the methyl group (4A), primary amine (4B), the alpha carbon of the glycine moiety (4C), and the methyl ether (4D).
Synthetic chemistry schemes
Figure 5 show the chemistry scheme used to make many of the 68 new analogues of F9000505 synthesized for this project.
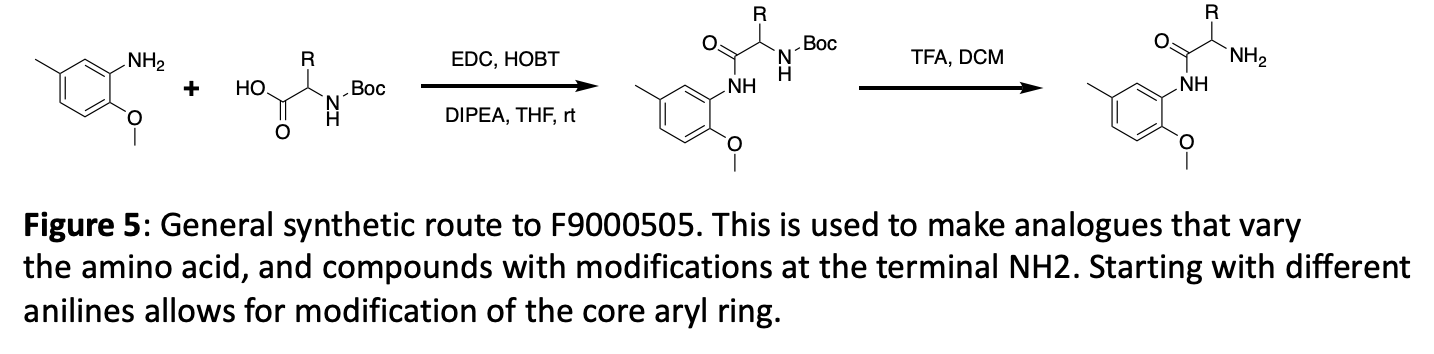
Figure 6 demonstrates utilization of an easily synthesized key intermediate to explore the addition of new functionality at three different distances from the core, linked with an amide. If any offer enhanced potency, follow up will focus on modifications of the pendant aryl ring.
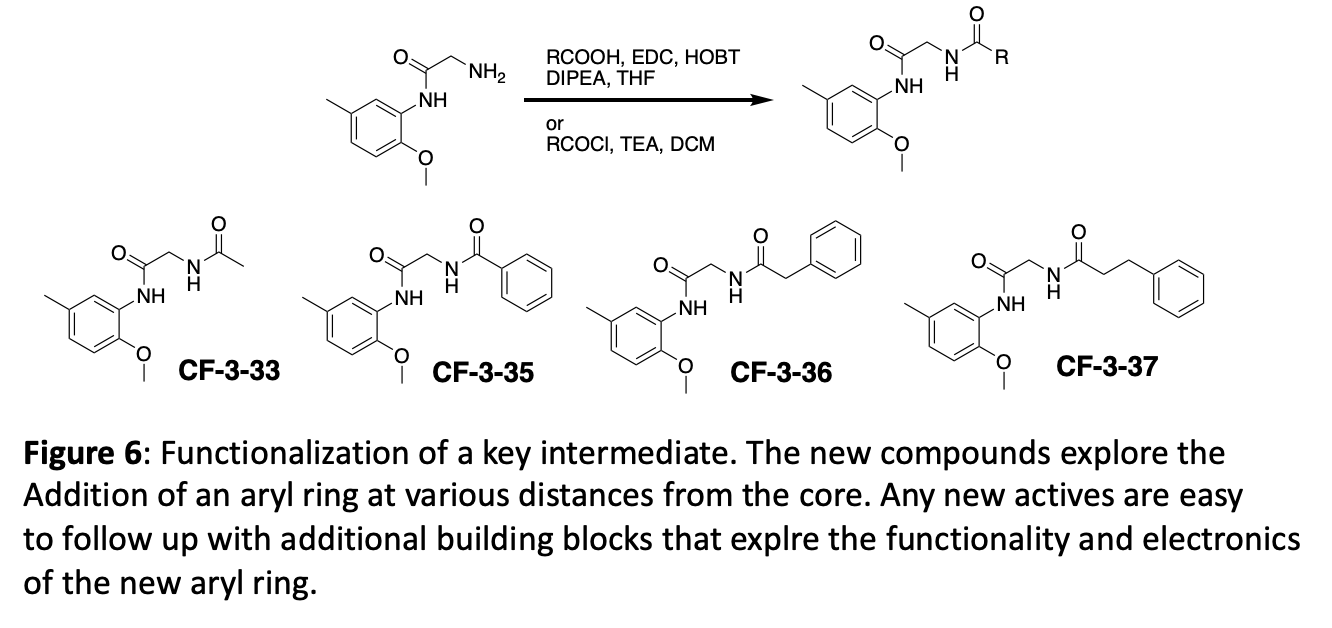
Figure 7 depicts three different ways to functionalize the same key intermediate with robust chemistry that provides different sets of products. A key element of the initial fragment analogue strategy is to generate and use key intermediates that can be diversified in the last synthetic step.
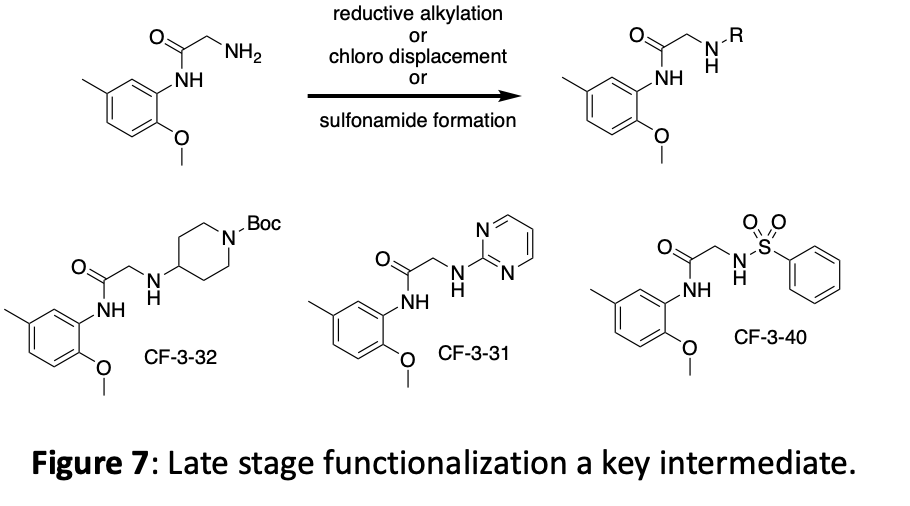
The crystal structure suggests that new interactions may be possible if we grow the molecule in the ether position. In addition to making several different ethers, we have opted to incorporate both an alkyne linker and a triazole linker in this position. These choices offer different spacing, different angles, different functionalities, and can be expanded with robust chemistry. An overview of this linker modification chemistry is shown in figure 8.
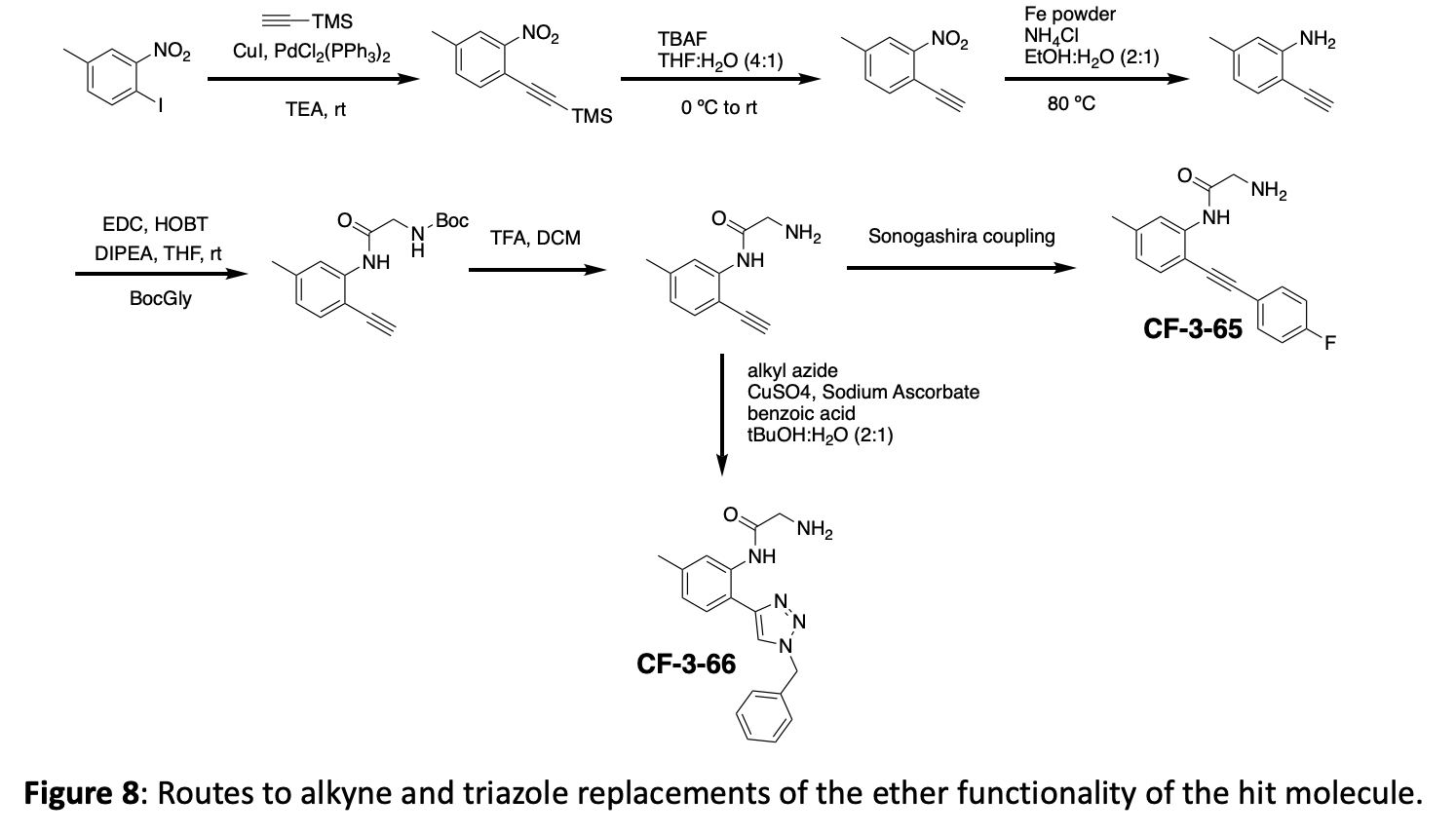
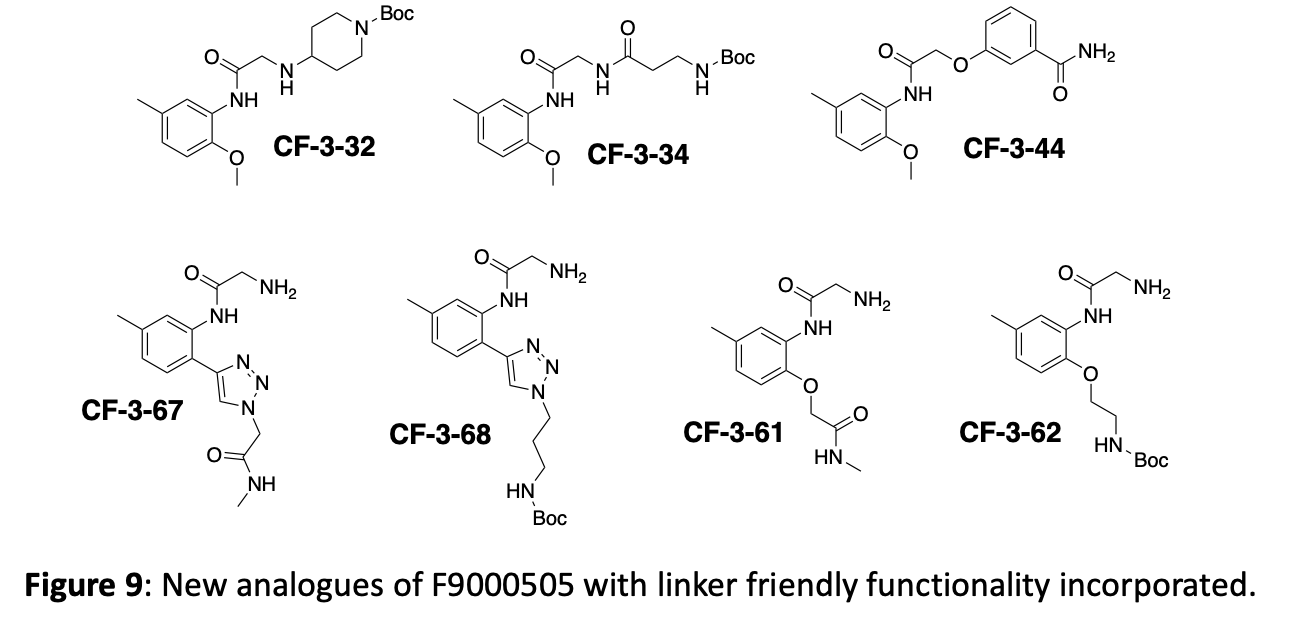
Finally, a key element of the strategy is to identify positions on the molecules where linkers can be attached to generate bifunctional degradation reagents. Crystallography will of course provide critical information to guide linker location. To improve our chances of quickly identifying potential linker locations, we are routinely incorporating some building blocks with functionality that lends itself to linker friendly chemistry. Currently these are mostly simple amides and Boc-protected amines, but azides and alkynes would also be valuable additions. Some newly synthesized compounds with “linker-friendly” functionality are shown in figure 9.
Stay tuned for details on compounds we have made and purchased for other sites and, importantly, the SPR screening data that should be available soon that lets us know how we are doing in terms of binding affinity!
Don’t hesitate to reach out with questions and comments.