Since we posted our analysis of genetic variation of SARS-CoV-2 main protease (MPro) with inhibitor N3 (PDB: 7bqy), there have been other inhibitors co-crystallized with the MPro (PDB: 6lze, PDB: 6m0k, and PDB: 6y2f).
In my last post, I talked about the two inhibitors, 11a and 11b (PDB: 6lze, PDB: 6m0k) and in today’s post, we focus on another MPro structure co-crystallized with an inhibitor called 13b (PDB: 6y2f).
The premise for the MPro inhibitor discussed in this post is based on the authors’ previous work where they designed a peptidomimetic α-ketoamide, 11r (Figure 1), as a broad-spectrum inhibitor of coronavirus and enterovirus replication. (1) In a subsequent study (2), the authors attempt to improve the half-life of the previous inhibitor in plasma by incorporating the p3-p2 amide bond in 11r into a pyridine ring in 13a. (Figure 1) They speculated that this change might hide the amide bond from cellular proteases and hence prevent its cleavage. Furthermore, the authors justified that to enhance the antiviral activity against betacoronaviruses of clade b (SARS-CoV-2 and SARS-CoV) they replaced the cyclohexyl moiety of 13a by cyclopropyl in 13b which is a smaller moiety. By changing 13a to 13b, they sacrificed the goal of broad-spectrum inhibitor activity of their compound, as the cyclohexyl in 13a was intended to fill in the enterovirus MPro which is replaced by a smaller cyclopropyl moiety in 13b.
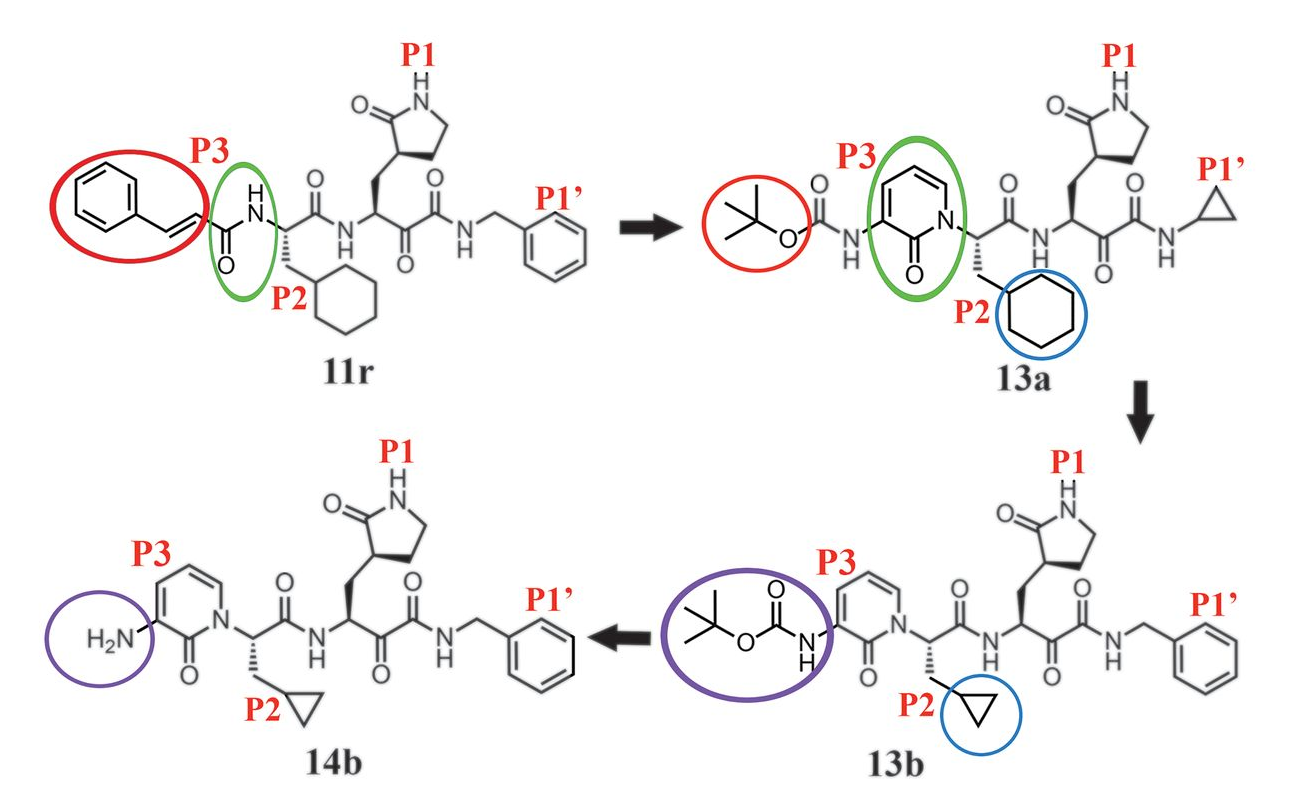
Figure 1. The design rationale for an improved alpha-ketoamide inhibitor, the path from 11r to 13b.
The IC50 value of 13b was at 0.67 ± 0.18 μM for SARS-CoV-2 MPro and 0.90 ± 0.29 μM and 0.58 ± 0.22 μM for SARS-CoV Mpro and the MERS-CoV Mpro respectively. Additionally, the researchers in this study showed that 13b has lung tropism which means that 13b was detectable in mice’s lung tissue 4 hours following its subcutaneous administration. Lung tropism is desirable considering the tissue that COVID-19 mostly affects.
The catalytic residue of the MPro catalytic site, Cys145, performs a nucleophilic attack onto the α-keto group of 13b to form a thiohemiketal bond in a reversible reaction and thus MPro is inhibited.
Their study provided a framework for the development of pyridone-containing inhibitors to be used as antiviral drugs.
In this post, I will explain how the non-synonymous variants found at SARS-CoV-2 MPro catalytic site across thousands of SARS-CoV-2 samples from COVID-19 patients are predicted to affect 13b binding. (3)
To predict the effect of those identified mutations on inhibitor (13b) binding, we use ICM software (Molsoft, SD). In ICM, by mutating the residues, we can see the change in Gibbs free energy of inhibitor binding (ddGbind) which would indicate if the mutation penalizes the binding of the inhibitor significantly or not.
To see the detailed methods on how the change in Gibbs free energy of ligand binding is calculated, please view this Zenodo post here. Below in table 1, we show the four non-synonymous mutations and the corresponding ddGbind values.
| Residue number | Wild Type | Mutant | ddGbind | dGbind_wild | dGbind_mut |
| 49 | Met | Ile | -0.6 | -27.1 | -27.7 |
| 52 | Pro | Ser | -0.1 | -27.1 | -27.2 |
| 142 | Asn | Ser | 1.7 | -27.1 | -25.4 |
| 168 | Pro | Ser | 0.4 | -27.1 | -26.7 |
Table 1. The predicted change in Gibbs free energy associated with the non-synonymous variants found at MPro catalytic site across more than 15000 SARS-CoV-2 samples. ddGbind values are in Kcal/mol.
Based on the precision of our method, none of the non-synonymous mutations are predicted to affect 13b binding significantly, since all ddGbind values are less than 2.0 Kcal/mol. (4) Prediction of no significant changes in ddGbind values for the four non-synonymous mutations (M49I, P52S, N142S, and P168S) is possibly due to the conservation of key ligand-protein interactions after these residues are mutated. In figure 2, we show the 2D ligand-protein interaction diagram which outlines the chemical interactions between 13b and MPro catalytic site residues.
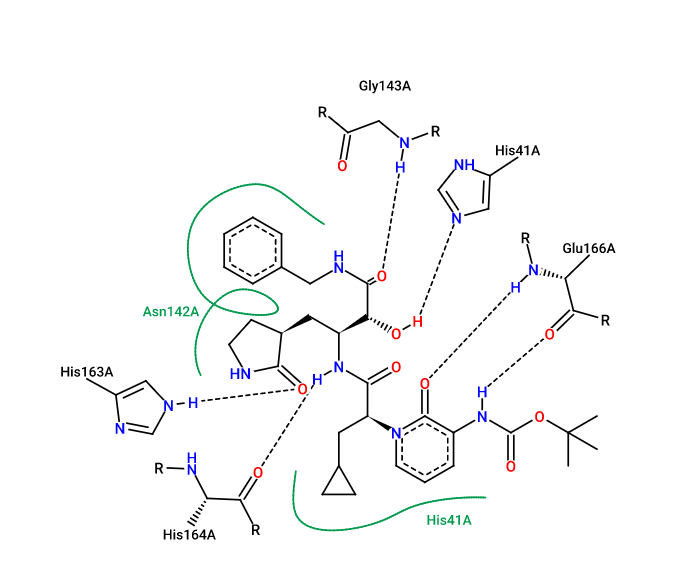
Figure 2. The 2D ligand-protein interaction diagram between ligand 13b and the residues of the MPro active site. (PDB: 6y2f). This diagram was created using Pose.View in Proteins.Plus.
We created an energy matrix table where we consider the effect of every possible mutation on the binding of the ligand (inhibitor 13b) with SARS-CoV-2-MPro. Table 2 includes a list of the 20 residues at MPro catalytic site expect Cys145. Cys145 is not included as it makes a covalent bond with 13b: any mutation will severely affect 13b binding.
It is likely that the residues of the rows with green color are not making optimal interactions with 13b. This is reflected in the very slight and non-significant lowering in ddGbind values of these residues (mutations are stabilizing) and hence the green color. On the other hand, the sidechains whose mutations are shown in orange are likely the positions where MPro would better interact with 13b which include H41, S144, M165, E166, P168, D187. As a result, the mutations of those sidechains are predicted to penalize inhibitor-binding significantly and hence the orange color. According to Zhang and her colleagues (3), His41 provides a hydrogen bond to stabilize the oxyanion group of thiohemiketal link between 13b which is also reflected in the orange row in table 3. The amide oxygen of 13b partly accepts a hydrogen bond from the main-chain amide of Ser144, which forms the canonical “oxyanion hole” of the cysteine protease, making S144 an important residue for 13b inhibitory mechanism.
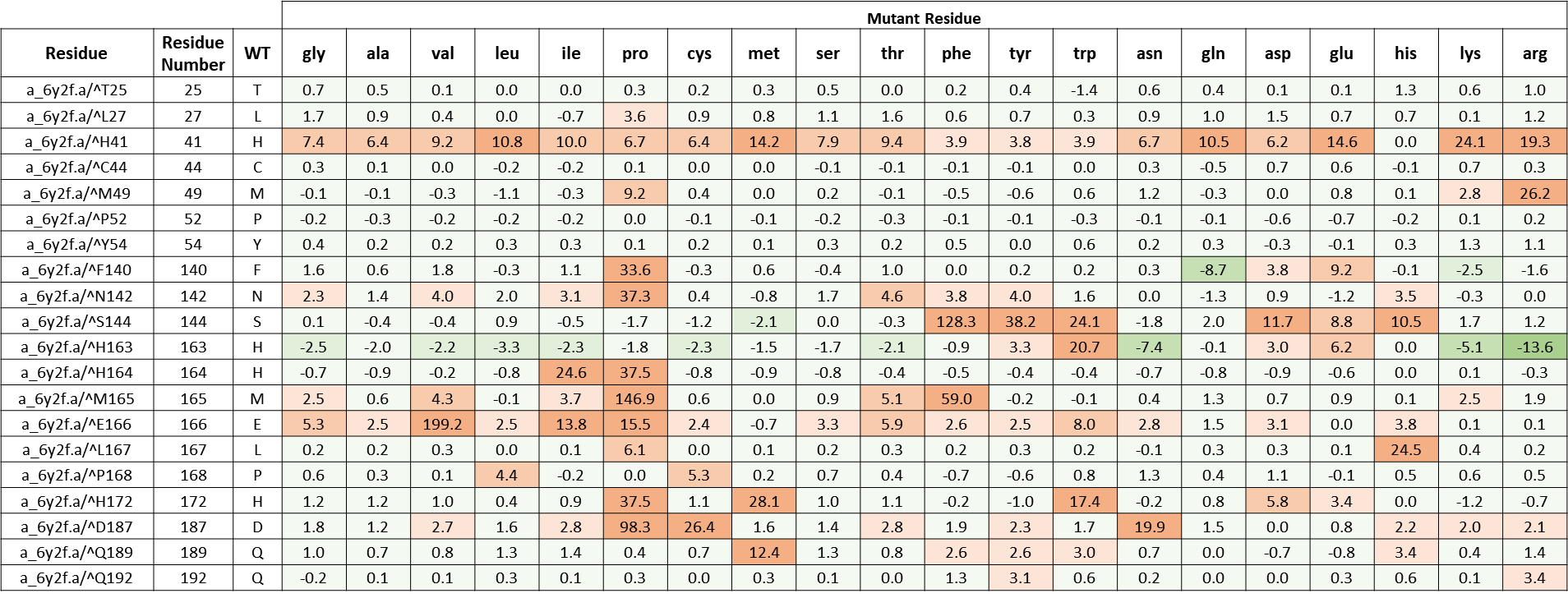
Table 2. The energy matrix showing the effect of all possible mutations at the SARS-COV-2 MPro catalytic site and the predicted effect on inhibitor 13b binding. No mutations at Cys145 were considered since it makes the covalent thiohemiketal linkage with 13b which is essential for the inhibitory mechanism in this scenario. The energy unit is kcal/mol.
Altogether, for the improved α-ketoamide inhibitor, 13b, we can conclude that the rare variants found in SARS-CoV-2 samples are only expected to minimally affect 13b binding to the MPro.
References:
- Zhang, Linlin., α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: Structure-based design, synthesis, and activity assessment. Journal of Medicinal Chemistry. 63 (9), 4562–4578 (2020). https://doi.org/10.1021/acs.jmedchem.9b01828
- Zhang, Linlin., Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368(6489), 409-412 (2020). https://doi.org/10.1126/science.abb3405
- Nicola De Maio. Genetic diversity at the catalytic site of the main SARS-Cov-2 protease. (2020) openlabnotebooks.org –https://openlabnotebooks.org/genetic-diversity-at-the-catalytic-site-of-the-main-sars-cov-2-protease/
- Schapira, M., Totrov, M., Abagyan, R. Prediction of the Binding Energy for Small Molecules, Peptides and Proteins. Journal of Molecular Recognition 12(3), 177-90 (1999). https://doi.org/10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z