Written in collaboration with Tigran M. Abramyan (Alexander Tropha’s lab at UNC Chapel Hill)
In my previous open lab notebook entry I described the in silico approach using the program SparkTM to grow fragments found to co-crystallize within binding site A of the transcription factor brachyury to compounds with hopefully higher binding potency. The overall aim of this project is the development of modulators of brachyury as well as the exploration of brachyury’s role in chordoma and intervening strategies. As mentioned in the previous entry brachyury (TBXT) is a transcription factor that is implicated in a rare type of cancerous tumor named chordoma affecting 300 Americans annually.[1-3]
As a quick reminder, the group at SGC Oxford headed by Opher Gileadi performed crystal soaking experiments of both wild type and G177D mutant of brachyury protein with a poised library of 768 diverse fragments[4,5] and showed that 29 fragments bound to five pockets on wild type brachyury labeled A-E and one pocket on the mutant labeled F (cf. Fig. 1A, B).
Since the SparkTM approach is not applicable to the fragments in pocket B and F due to the large overlay of the fragments we initiated a collaboration with Alexander Tropsha’s group from the department of pharmacoinformatics at the UNC Eshelman School of Pharmacy. I worked closely with Tigran Abramyan and Hatice Gokcan to use different in silico approaches for the development of new compounds.
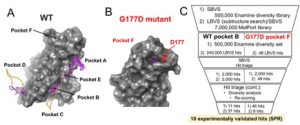
Figure 1. (A) Pockets A-E of wild type brachyury mapped by 27 fragments (magenta sticks) in crystal soaking experiments (see text for more details). (B) Pocket F of G177D mutant protein mapped by two fragments. (C) Virtual screening workflow used to select the hit molecules for purchase. The validation of the virtual screening hits using surface plasmon resonance binding analysis method determined ten molecules as experimental hits (binders).
We explored the structure of the target protein, analyzed the modes of binding of the fragments and assessed the binding sites as well as the impact of crystal packing on the ligand binding.[6] Through these analyses we determined that the crystal contacts played a major role in fragment binding by shaping artificial pockets at the protein-protein interface which otherwise are absent in the single protein. This is especially true for pocket A. Two pockets, pocket B in wild type and pocket F in G177D mutant, were subsequently designated as ‘true’ pockets and were further pursued in drug design. Encouragingly, the presence of the mutant D177 residue in pocket F of the mutant bares the potential to develop fragments bound to pocket F into mutant-selective inhibitors.
Using the two pockets virtual screening (high-throughput molecular docking) was performed in search for virtual hit molecules to then be experimentally validated. The schematics of our virtual screening pipeline is shown in Figure 1C. In order to increase the likelihood of success in our virtual screening studies we first intended to validate our docking methodology by developing a docking model that would be able to reproduce the available experimental observations. Since the only available experimental data were the 3D coordinates of the soaked fragment-protein complexes, we developed a docking model that was able to correctly predict the binding modes of the fragments in the crystal structures (with RMSDs with respect to the crystal structure < 1.5 Å). Additionally, we performed cross-docking of all 29 fragments and showed that in our model the fragments preferred their crystal pockets over the other pockets. This validated docking model was then used for virtual screening.
Two sources of commercially available compound libraries were used for virtual screening: MolPort (7.6 million compounds) and Enamine 500k diversity set (which represents 2.6 million ready to screen Enamine compounds). MolPort was filtered using the drug-likeness rules in Pipeline Pilot (Discovery Studio) which resulted in ˜5.6 million molecules. We then used the fragments of wild type’s pocket B and G177D mutant’s pocket F and their analogues together with the prepared MolPort library to conduct ligand-based virtual screening (LBVS). To this end, substructure search was performed using the fragments and their analogs as substructures, which resulted in ˜240,000 hits for pocket B and 48 hits for pocket F. These hits were subsequently used in structure-based virtual screening (SBVS) where molecular docking simulations in Glide available through Maestro (Schrödinger Release 2018-4: Glide, Schrödinger, LLC, New York, NY, 2018) were performed in order to prioritize the molecules to be purchased by sorting with Glide docking score.[7] In order to ensure chemical diversity of our hits we performed an independent SBVS using the aforementioned prepared Enamine 500k diversity set library.
For each pocket from each VS approach (the LBVS-SBVS combination and the separate SBVS), 2,000 hits based on the docking scores were selected for further re-scoring and hit triage. The re-scoring was performed with the MMGBSA scoring function available in Maestro by considering molecular mechanics minimization of the binding site amino-acid residues (defined as the residues within 5Å from the ligand). During the hit triage which was the selection of the final molecules for experimental validation the common steps in both VS approaches were: visual inspection to ensure the molecules were well-buried in the pockets and made at least three bonds with the protein which were present in the fragment-protein crystal structures; the selection of a representative molecule from a cluster after clustering; and selection of the molecules with both high Glide docking score and MMGBSA score (higher than those of the crystal fragments). The first visual inspection of 2,000 hits of each VS approach was performed by Tigran Abramyan, Hatice Gokcan and me and the further limitation of the number of compounds was conducted together with the PI’s of the Willson and Tropsha group. In the LBVS-SBVS approach it was ensured that the ‘parent’ fragment within the larger molecule had the same binding mode as in the crystal structure of the fragment-protein complex.
To date the first round of experimental validation has been completed by Joseph Newman in the SGC Oxford using surface plasmon resonance (SPR) binding analysis, DSF screening and X-ray crystallography. Ten virtual screening hits bind the protein and compound Molport-039-308-236 revealed the lowest Kd of 26 µM in the SPR assay. Encouragingly SGC resolved a crystal structure of the protein in complex with MolPort-020-049-143 whose binding pose fits well to the docked pose from virtual screening with RMSD=1.03 Å (cf. Fig. 2) although the compound displays a relatively low ΔTm of -0.11°C in the DSF assay.
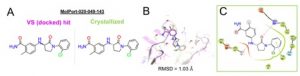
Figure 2. (A, B) The brachyury bound crystal structure of MolPort-020-049-143 obtained from our virtual screening reproduces the virtual binding mode of the compound docked on pocket F of G177D brachyury. VS hit and the co-crystallized compound represent each one enantiomer of the racemic mixture MolPort-020-049-143. (C) Ligand interaction diagram of the compound crystallized in crystal soaking experiments.
These preliminary results for a new protein with no well-defined binding sites are encouraging. The relatively low binding affinity of the best hit to date is likely due to the lack of well-defined small molecule binding sites on the protein. SGC Oxford aims to repeat the ligand-protein binding assessments and crystallization experiments for validation. The 3D structures of the best binders will further guide hit-to-lead optimization steps in brachyury inhibitor design. Moreover, because only the cluster representatives were selected for purchase during the hit triage, for the experimentally validated hits we aim to expand the respective clusters and purchase molecules similar to the hits from the clusters for the next round of experimental validation, in attempts to increase the binding affinity of the initial hits. Upon the discovery of the final hit molecules medicinal chemistry optimization through SAR will be performed. The results of the brachyury hit discovery efforts will be prepared for a peer-reviewed publication.
References
- Sharifnia T, Wawer MJ, Chen T, Huang QY, Weir BA, Sizemore A, et al. Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat Med. 2019;25(2):292-300.
- Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet. 2012;44(11):1185-7.
- Bosotti R, Magnaghi P, Di Bella S, Cozzi L, Cusi C, Bozzi F, et al. Establishment and genomic characterization of the new chordoma cell line Chor-IN-1. Sci Rep. 2017;7(1):9226.
- Scherer MK, Trendelkamp-Schroer B, Paul F, Perez-Hernandez G, Hoffmann M, Plattner N, et al. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J Chem Theory Comput. 2015;11(11):5525-42.
- Cox OB, Krojer T, Collins P, Monteiro O, Talon R, Bradley A, et al. A poised fragment library enables rapid synthetic expansion yielding the first reported inhibitors of PHIP(2), an atypical bromodomain. Chem Sci. 2016;7(3):2322-30.
- Jacobson MP, Friesner RA, Xiang Z, Honig B. On the role of the crystal environment in determining protein side-chain conformations. J Mol Biol. 2002;320(3):597-608.
- Friesner R A, Murphy R B, Repasky M P, Frye L L, Greenwood J R, Halgren T A, Sanschagrin P C, Mainz D T. Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J Med Chem. 2006;49(21):6177-6196.
Nice work. Your application of virtual screening to convert weak fragment hits into micromolar leads is an innovative approach to a very challenging problem. The workflow you outline could be applied to a wide range of proteins where fragment structures have been obtained by the SGC.