Every cell in your body contains the exact same set of genetic instructions encoded in its genome. However, for complex multicellular organisms such as ourselves, we depend on many specialized cell types that exhibit strikingly different features. This is largely accomplished by precisely controlling how and when genetic information is read out in each cell, known as a cell’s gene expression profile or transcriptional program. Cancer results from mutations, which drive abnormal transcriptional programs that ultimately manifest as the hallmarks of cancer, including evasion of cell death and unlimited replicative potential. If we can learn more about how these transcriptional programs are executed both in health and disease, we may be able to find ways to rewire dangerous gene expression patterns to either destroy cancer cells or force them into a benign state.
One way in which transcriptional programs are enforced is through distal tissue-specific enhancer regions that activate gene expression in specific cell types (Figure 1). Enhancer regions also become co-opted in some cancers, resulting in increased production of oncogenes (genes that promote cancer) [1]. Cancer cells often become dependent on the expression of these genes to maintain tumour growth, a feature that is frequently referred to as oncogene addiction. If we can find ways to disrupt the enhancer elements that are driving expression of these genes, we may be able to turn off oncogenic transcription programs. My research focuses on the components that mediate enhancer biology and I hope this work will expand our understanding of the regulation and misregulation of transcriptional programs.
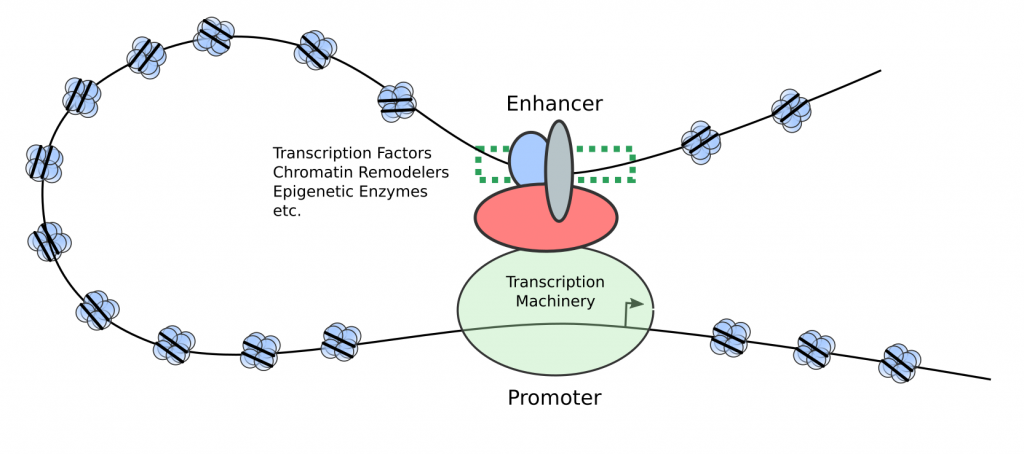
Figure 1 – Enhancers are distal genetic elements that become bound by transcriptional activators to promote the transcription of a gene with the potential to influence cell identity.
For this project, which I will be sharing my data prepublication through this blog, I will focus primarily on a novel regulator of enhancer function called NSD3. In a recent publication, Chen et al found that NSD3 is part of a protein complex that promotes enhancer activity and is required for the maintenance of acute myeloid leukemia (AML), a cancer of the blood [2]. The authours of this study found that a shortened version (isoform) of NSD3, which lacks several functional regions of NSD3, including its histone methyltransferase domain, is responsible for mediating an interaction between the proteins BRD4 and CHD8, two components that are critical for enhancer function. The short isoform also turns out to be the most dominant form present in AML patient samples described in the public TCGA database (Figure 2). Importantly, further research is needed to determine NSD3’s exact role at enhancers and whether it is a suitable drug target for the treatment of AML and other cancers. To address this problem, I will use bioinformatic methods to mine publically available datasets as well as perform cell biology experiments to further explore how NSD3 contributes to enhancer activity in cancer. All data for this project will be hosted on Zenodo with links, updates, and brief literature reviews relevant to the work posted to this blog.
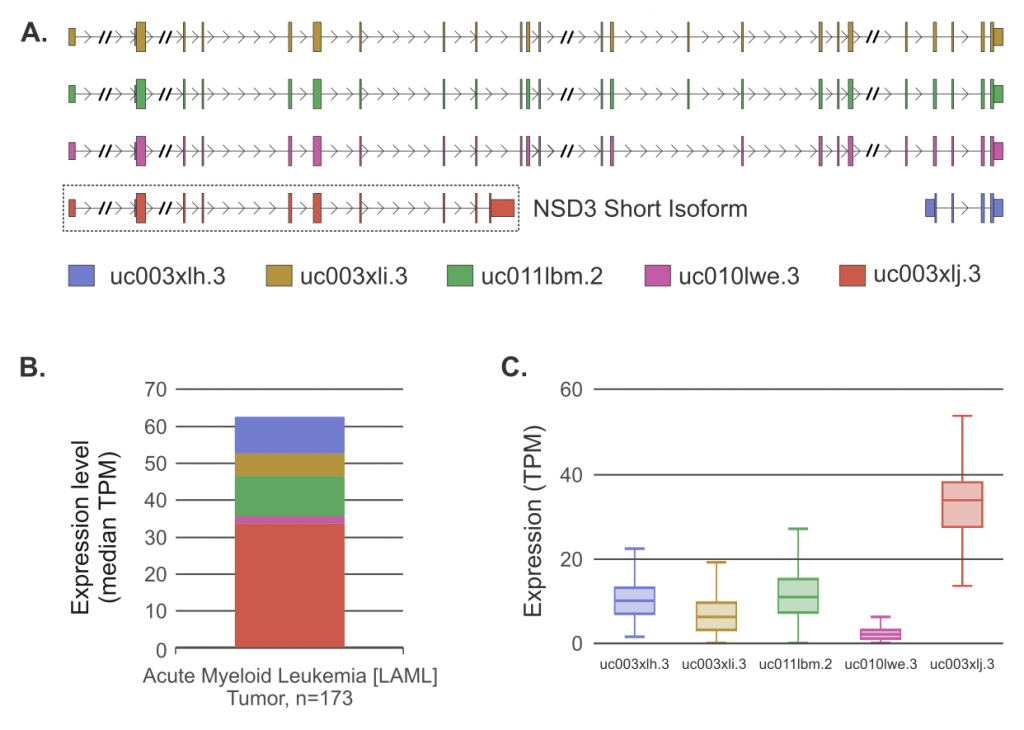
Figure 2 – (A) NSD3 isoforms annotations from UCSC genome browser. The short isoform is highlighted. Intron lengths have been shortened where indicated (//) for clarity. (B) Median RNA-Seq expression levels in Transcripts per Million Reads (TPM) for all NSD3 isoforms across 173 TCGA-LAML patient samples obtained from ISOexpresso platform [3]. (C) Box plot of NSD3 isoform expression (TPM) from TCGA-LAML dataset. The short NSD3 isoform is the most dominant.