Scaffold hopping is a strategy utilized in medicinal chemistry where the core chemical structure of a promising drug candidate is replaced isosterically, which leads to structurally novel compounds. There are many reasons for undertaking this exercise including; improving potency, changing physiochemical properties such as solubility and clogP, enhance ADME properties and to move into new intellectual property space. Figure 1 is an example of scaffold hopping where potency was greatly improved by switching the heteroatom from oxygen to sulfur and changing its position on the ring.
 Figure 1: A scaffold hop generates a compound with increased potency.
Figure 1: A scaffold hop generates a compound with increased potency.
One promising benefit from scaffold hopping in the field of kinase inhibitors is that ‘small’ modifications within the hinge binding region can affect drastic changes to the kinome selectivity profile of the inhibitor. As part of the SGC, one of our goals is to develop chemical probes for kinases. Amongst the strict criteria for chemical probe declaration is selectivity, specifically >30-fold selectivity relative to other sequence-related proteins of the same target family. As previously discussed in a previous post, we have been investigating structures based on a published GSK compound, GSK650394 (Figure 2).
 Figure 2: GSK650394, an azaindole with potent CaMKK2 activity.
Figure 2: GSK650394, an azaindole with potent CaMKK2 activity.
GSK650394 is an extremely potent inhibitor of CaMKK2, with single digit nanomolar IC50 in a biochemical assay. When selectivity data was collected it showed the compound to be extremely promiscuous. In a screen of 300 kinases, 31 were inhibited by >90% at 1 µM, excluding GSK650394 from ever being used as a chemical probe to investigate CaMKK2. The azaindole scaffold is commonly found in kinase inhibitors and is often referred to as a ‘privileged’ structure, reflective of its extensive use in the field of medicinal chemistry and drug discovery programs.
Encouraged by the GSK650394’s potency, we decided to undertake a scaffold hopping program around the azaindole core. We decided we would retain the auxiliary R-groups, but also make reductionist analogues without the phenyl substituent (Figure 3). The goal of this strategy we to find equally potent novel hinge binders with a greatly improved selectivity profile.
 Figure 3: Schematic representation of our strategy in scaffold hopping.
Figure 3: Schematic representation of our strategy in scaffold hopping.
Our synthetic efforts focused on creating matched-pair compounds that had a varied topographical relationship to the parent, increasing ring heteroatom counts, modulation of hinge binding interactions and changing from fused rings to rings connected via rotatable bonds. GSK650394 has two hydrogen bonding interactions with the hinge region of CaMKK2. Our first set of compounds were designed to probe how modifications to these interactions are tolerated. Modifications include removing the H-bond donor by substitution with another heterocycle, such as sulfur or oxygen, or by using the N-methyl azaindole analogue (Figure 4). The other analogues shown in Figure 4 will change both H-bond interactions and incorporate increasing heterocyclic character. Although these look very similar, the changes in bond lengths due around the ring will cause each auxiliary group to occupy different space. For clarity in the proceeding figures, only the phenyl substituted of the matched pair is shown.

Figure 4: Scaffold hops that retain the fused 6,5-membered ring system with 3,5-substitution patterns.
The next set we designed was also based on a fused 5,6-membered ring systems, but with substitutions at the 2,4- positions rather than the 3,5- positions, as in the parent compound and above analogues are (Figure 5). We incorporated molecules that would allow us to elucidate the role of H-bonding to the hinge again, with H-bond acceptors, H-bond donators and non-bonding carbons being included.

Figure 5: Scaffold hops retaining the fused 6,5-membered ring system but with 2,4-substitution patterns.
Our next set of isosteres consisted of fused 6,6-membered ring systems of the quinoline and quinazoline core (Figure 6). These molecules offer only 1 possible hinge-binding interaction. The 2-methyl quinoline analogue is an interesting molecule. Some kinases can tolerate a methyl in close vicinity to the hydrogen bonding region of the inhibitor, while in others it will incur a severe penalty to it in potency. This means under the right circumstances, with the right kinases and inhibitor combination, a methyl alpha to the hydrogen bond can lead to a greatly enhanced selectivity profile. The set designed can potentially teach us about CaMKK2s ability to tolerate this scenario. If both quinoline and 2-methyl quinoline are active, a KINOMEscan (DiscoverX) would elucidate the effect of the methyl in the 2 position.
 Figure 6: Quinoline and quinazoline isosteres of the azaindole.
Figure 6: Quinoline and quinazoline isosteres of the azaindole.
The last set of compounds is the furthest removed from the azaindole core. They are based on the pyrimidine core and we designed to have either one or two hydrogen bond interactions with the core (Figure 7). The (6-aminophenyl)-pyrimidine derivative is very similar to a potent compound in the original publication. We will learn about the effects of having the hydrogen bond donor in the compounds and whether it is an essential interaction for activity.
 Figure 7: Pyrimidine based derivatives representing more drastic hops from the original scaffold.
Figure 7: Pyrimidine based derivatives representing more drastic hops from the original scaffold.
A total of 16 matched pairs, giving a total of 32 compounds, will be tested in a biochemical assay for CaMKK2 activity. The small library we created will allow us to test the tolerance of CaMKK2 to small changes, alongside more considerable changes.
An insightful tool to compare the library, or to be preemptively used in designing varied libraries, is SwissADME. It is a free software provided by the Swiss Institute of Bioinformatics, described as a ‘tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules’. It is extremely useful and provides analysis/predictions on physiochemical properties, lipophilicity, solubility and other useful parameters. An example of the analysis is shown on some of the compounds made of this library in Figure 8. Our team often utilizes this software for analysis across libraries of compounds.
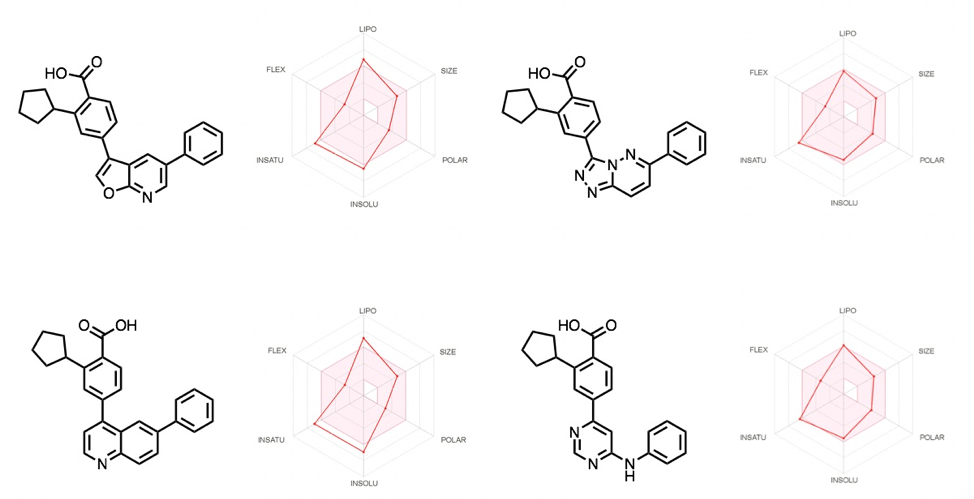 Figure 8: A selection of compounds analyzed using Swiss ADME software.
Figure 8: A selection of compounds analyzed using Swiss ADME software.
I will write a follow up post once the full biological data has been collected. The preliminary results look very good (Percentage of control at 1 µM), with some compounds having potent inhibition.
The chemdraw file associated with this post can be downloaded here… Download SDF file
