Frances M. Potjewyd and Vittorio Katis
http://doi.org/10.5281/zenodo.4670214
Spleen Tyrosine Kinase (SYK) has a reported role in AD pathology and was identified as a target for the TREAT-AD (Target Enablement to Accelerate Therapy Development for Alzheimer’s Disease) program.1–3 We are focusing drug discovery efforts on the inhibition of SYK and more specifically, inhibition of the interaction of SYK SH2 domains with receptor FCεR1γ. With the generation of novel chemical tools, we hope to delineate the effect on inhibition of this kinase and receptor interaction on aspects of AD pathology.
The mechanisms of SYK activation include auto-phosphorylation and binding to phosphorylated ITAM present at receptors such as FCεR1γ which results in phosphorylation by proteins such as LynB.4 Activated SYK directly or indirectly phosphorylates downstream targets which have intricate roles in AD pathology.5 Hall et al describe the mechanism of inhibition of an allosteric SYK inhibitor, X1, which is believed to act by keeping SYK in an inactivated and contracted state.6 They report SYK activation by ITAM binding and phosphorylation causes expansion of a solution structure of SYK. X1 inhibits activation of SYK via phosphorylation (auto- and via LynB) and through ITAM binding (X1 is non-competitive with ITAM binding).
There are no further literature reports of this compound, and to our knowledge there is no available synthetic route, therefore, we designed a synthetic route. Synthesis (Figure 1.): The cyclopropylamine is installed on the anyhydride (1) by heating the reagents in acetic acid in the presence of sodium acetate. The addition of water to the reaction leads to precipitation of (2) in good yields. The fluoro-intermediate (2) is heated with methyl 4-hydroxybenzoate in a sealed microwave vial with potassium carbonate in DMF to yield (3). (3) is subjected to ester hydrolysis with lithium hydroxide in water and THF to yield a mixture of two ring opened products (4a–b). Ring closure to generate X1 was achieved with thionyl chloride in toluene, followed by an acidic aqueous work-up. Acid hydrolysis would likely prevent ring opening, however intermediates (4a–b) could also be tested as analogues of X1 for allosteric SYK inhibition.
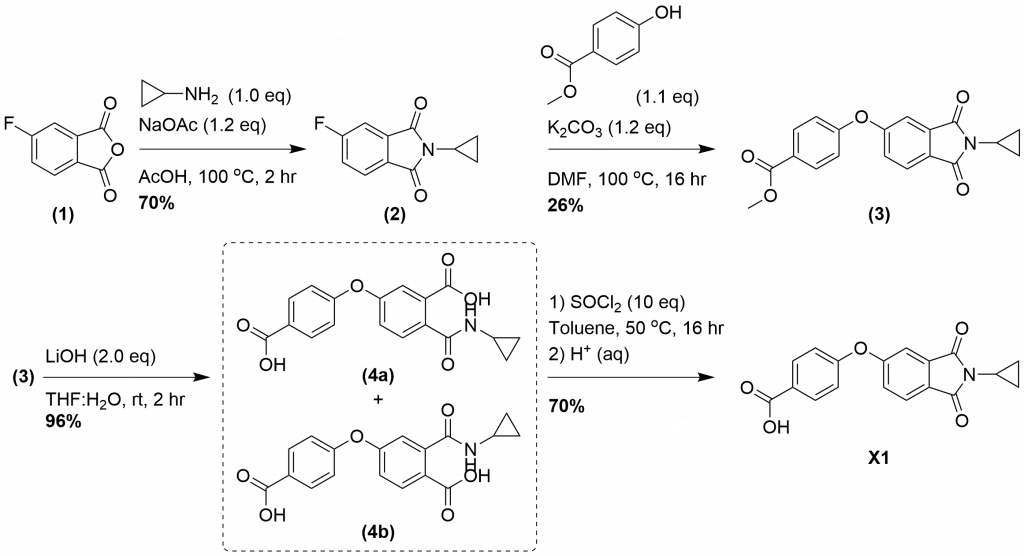
Figure 1. Synthetic scheme for the synthesis of allosteric SYK inhibitor X1.
Due to the ring opening occurring within the synthesis we hypothesized there could be a covalent mode of inhibition. Compounds (3), (4a–b), and X1 were tested via intact mass analysis (Quadrupole time of flight – QTOF) with the tandem-SH2 domains of SYK (tSH2-SYK) at The University of Oxford by Vito Katis (Figure 2A.). These data show a mass adduct of +284.29 at greater than 50% labelling and 568.66 as a minor adduct. Intriguingly, this corresponds to a mass which infers loss of the cyclopropyl group (Figure 2B). Further to this, there are four cysteines present in tSH2-SYK that are shown to be reactive to covalent SYK inhibitors such as BBL010720 (a covalent inhibitor with competitive with p-ITAM binding to SYK).7 This suggests that X1 is able to bind to tSH2-SYK with a degree of selectivity.
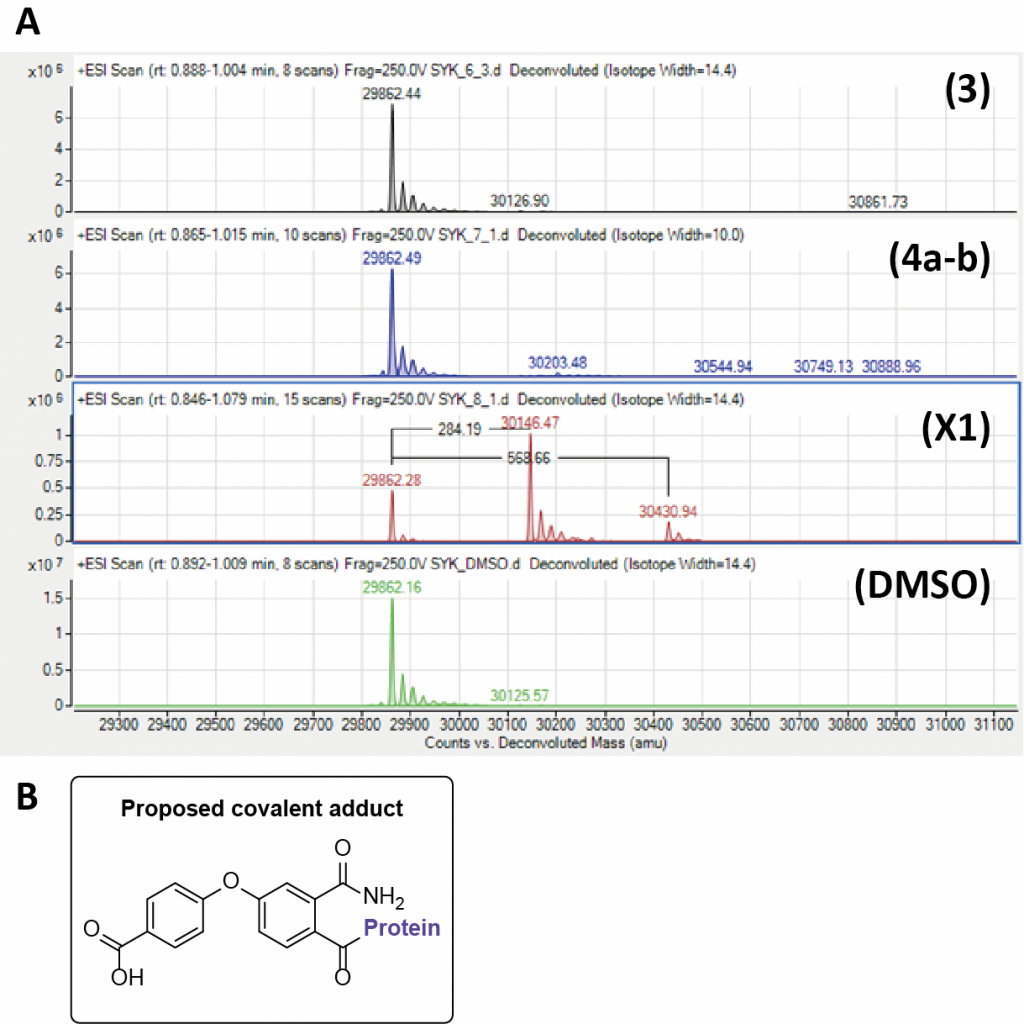
Figure 2. A) Intact mass analysis of allosteric inhibitors with the tandem-SH2 domains of SYK (tSH2-SYK) B) The proposed covalent adduct of X1.
We plan to explore this mechanism of inhibition by synthesizing a variety of analogues that alter the chemical components of the molecule to begin a structure-activity relationship study, and to probe if the covalent adduct is reliant on the cyclopropyl group present in X1.
Chemistry Experimental
2 – 2-cyclopropyl-5-fluoroisoindoline-1,3-dione: To a flask equipped with a reflux condenser was added 5-fluoroisobenzofuran-1,3-dione (1.0 g, 1.0 equiv, 6.0 mmol), cyclopropanamine (0.42 mL, 1.0 equiv, 6.0 mmol), sodium acetate (0.59 g, 1.2 equiv, 7.2 mmol), and acetic acid (10 mL, 0.17 mol) and the reaction heated to 100 °C for 3 hrs. The reaction was cooled, diluted with H2O, and filtered under reduced pressure to yield the desired product as a pale purple solid (889 mg, 70 %). 1H NMR (400 MHz, DMSO-d6): δ 7.89 (dd, J = 4.63, 8.22 Hz, 1H), 7.70 (dd, J = 2.26, 7.49 Hz, 1H), 7.63 (ddd, J = 2.30, 8.42, 10.48 Hz, 1H), 2.69 – 2.61 (m, 1H), 0.93 – 0.83 (m, 4H). 13C NMR (101 MHz, DMSO-d6): δ 167.4, 167.09, 167.06, 167.0, 164.5, 134.5, 134.4, 127.63, 127.61, 125.6, 125.5, 121.2, 120.9, 110.8, 110.5, 20.6, 4.9.
3 – methyl 4-((2-cyclopropyl-1,3-dioxoisoindolin-5-yl)oxy)benzoate: To a microwave vial was added methyl 4-hydroxybenzoate (408 mg, 1.1 equiv, 2.7 mmol), 2-cyclopropyl-5-fluoroisoindoline-1,3-dione (500 mg, 1.0 equiv, 2.4 mmol) and K2CO3 (404 mg, 1.2 equiv, 2.9 mmol) in DMF (9.75 mL, 0.25M). The reaction vessel was sealed and heated to 90 °C for 16 hrs. The reaction was cooled, diluted with H2O and extracted with EtOAc (100 mL). The organic layer was washed with 1M NaOH (100 mL) and brine (3 × 100 mL). The organics were dried with sodium sulfate and concentrated in vacuo. The crude material was purified by column chromatography (silica, 0-100% EtOAc in hexane) to yield the desired product as a white solid (210 mg, 25.6 %). 1H NMR (400 MHz, CDCl3): δ 8.07 – 8.03 (m, 2H), 7.78 (d, J = 8.16 Hz, 1H), 7.34 (d, J = 1.98 Hz, 1H), 7.28 (dd, J = 2.21, 8.16 Hz, 1H), 7.08 – 7.03 (m, 2H), 3.90 (s, 3H), 2.71 – 2.63 (m, 1H), 1.02 – 0.94 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 168.2, 168.1, 166.2, 161.9, 159.4, 134.4, 132.2, 126.7, 126.5, 125.3, 123.8, 119.2, 113.0, 52.3, 21.1, 5.3. LCMS: expected mass for [M+H]+ (C19H16NO5), 338.10 m/z; found, 338 m/z.
4a/b – 5-(4-carboxyphenoxy)-2-(cyclopropylcarbamoyl)benzoic acid/ 4-(4-carboxyphenoxy)-2-(cyclopropylcarbamoyl)benzoic acid: To a flask was added methyl 4-((2-cyclopropyl-1,3-dioxoisoindolin-5-yl)oxy)benzoate (16 mg, 1.0 equiv, 48.6 µmol) and LiOH (2.5 mg, 2.1 equiv, 0.10 mmol) in THF (194 µL, 0.25M) and H2O (44 µL, 50.0 equiv, 2.43 mmol) and the reaction stirred at room temperature. To the reaction was added 1M HCl (50 mL) and the organics extracted with EtOAc (50 mL). The organics were washed with brine (50 mL), dried with sodium sulfate and concentrated in vacuo to yield the desired product as a mixture of ring opened products as a white solid (15 mg, 96%). Characterized as a mixture of ring opened products: 1H NMR (400 MHz, MeOD-d4): δ 8.09 – 7.99 (m, 2.3H), 7.54 (d, J = 2.47 Hz, 0.53H), 7.45 (d, J = 8.35 Hz, 0.58 H), 7.25 (dd, J = 2.52, 8.34 Hz, 0.60H), 7.16 – 7.09 (m, 1H), 7.07 (d, J = 8.82 Hz, 1H), 7.01 (d, J = 2.45 Hz, 0.34 H), 2.82 – 2.72 (m, 1H), 0.79 – 0.70 (m, 2H), 0.66 – 0.59 (m, 2H). 13C NMR (101 MHz, MeOD-d4): δ 173.7, 173.6, 169.1, 169.0, 168.5, 168.2, 161.9, 161.3, 160.6, 158.2, 142.5, 135.3, 134.1, 133.19, 133.16, 131.0, 127.9, 127.5, 125.7, 123.4, 121.7, 120.3, 119.8, 119.2, 119.1, 23.8, 6.3, 6.2. LCMS: expected mass for [M+H]+ (C18H16NO6), 342.09 m/z; found, 342 m/z.
X1 – 4-((2-cyclopropyl-1,3-dioxoisoindolin-5-yl)oxy)benzoic acid : To a flask was added 5-(4-carboxyphenoxy)-2-(cyclopropylcarbamoyl)benzoic acid (8 mg, 1.0 equiv, 0.02 mmol) and thionyl chloride (17 μL, 10.0 equiv, 0.2 mmol) in toluene (200 µL) and the reaction heated to 50 °C for 2 hrs. The reaction was cooled and 1M HCl (10 mL) was added, the organics were extracted with EtOAc (10 mL) and washed with brine (20 mL). The organics were dried with sodium sulfate and concentrated in vacuo to yield the desired product as a white solid (5 mg, 70 %). 1H NMR (400 MHz, CDCl3): δ 8.17 (d, J = 8.96 Hz, 2H), 7.86 (d, J = 8.21 Hz, 1H), 7.44 (d, J = 2.01 Hz, 1H), 7.36 (dd, J = 2.19, 8.13 Hz, 1H), 7.12 (d, J = 8.94 Hz, 2H), 2.74 – 2.68 (m, 1H), 1.05 – 0.98 (m, 4H). 13C NMR (101 MHz, CDCl3): δ 160.1, 167.9, 167.2, 161.9, 160.8, 134.7, 134.3, 129.2, 127.5, 125.6, 124.7, 118.9, 114.0, 21.3, 5.4. LCMS: expected mass for [M+H]+ (C18H14NO5), 324.08 m/z; found, 324 m/z.
Acknowledgements
This work was supported by the National Institutes on Aging TREAT-AD Center for Alzheimer’s Disease grant U54 AG065187.
References
(1) Schweig JE, Yao H, Beaulieu-Abdelahad D, Ait-Ghezala G, Mouzon B, Crawford F, Mullan M, Paris D. Alzheimer’s disease pathological lesions activate the spleen tyrosine kinase. Acta Neuropathol Commun. 2017, 5, 69. PMID: 28877763; PMCID: PMC5588676.
(2) Schweig JE, Yao H, Coppola K, Jin C, Crawford F, Mullan M, Paris D. Spleen tyrosine kinase (SYK) blocks autophagic Tau degradation in vitro and in vivo. J Biol Chem. 2019, 294, 13378-95. PMID: 31324720; PMCID: PMC6737214.
(3) Paris D, Ait-Ghezala G, Bachmeier C, Laco G, Beaulieu-Abdelahad D, Lin Y, Jin C, Crawford F, Mullan M. The spleen tyrosine kinase (Syk) regulates Alzheimer amyloid-β production and Tau hyperphosphorylation. J Biol Chem. 2014, 289, 33927-44. PMID: 25331948; PMCID: PMC4256331.
(4) Kraskouskaya D, Duodu E, Arpin CC, Gunning PT. Progress towards the development of SH2 domain inhibitors. Chem Soc Rev. 2013, 42, 3337-70. PMID: 23396540.
(5) Paris D, Ait-Ghezala G, Bachmeier C, Laco G, Beaulieu-Abdelahad D, Lin Y, Jin C, Crawford F, Mullan M. The spleen tyrosine kinase (Syk) regulates Alzheimer amyloid-β production and Tau hyperphosphorylation. J Biol Chem. 2014, 289, 33927-44. PMID: 25331948; PMCID: PMC4256331.
(6) Hall J, Aulabaugh A, Rajamohan F, Liu S, Kaila N, Wan ZK, Ryan M, Magyar R, Qiu X. Biophysical and mechanistic insights into novel allosteric inhibitor of spleen tyrosine kinase. J Biol Chem. 2012, 287, 7717-27. PMID: 22219190; PMCID: PMC3293529.
(7) Visperas PR, Winger JA, Horton TM, Shah NH, Aum DJ, Tao A, Barros T, Yan Q, Wilson CG, Arkin MR, Weiss A, Kuriyan J. Modification by covalent reaction or oxidation of cysteine residues in the tandem-SH2 domains of ZAP-70 and Syk can block phosphopeptide binding. Biochem J. 2015, 465, 149-61. PMID: 25287889; PMCID: PMC4410814.