Introduction
Situation
-
-
- Kinases are useful for drug discovery – many opportunities!
- Kinase by kinase probe development is important, but it will take too long to discover a probe for each kinase
- Disease relevant phenotypic assays need smart compounds sets
- Numerous high quality kinase inhibitors exist in the literature and in company collections
- Kinases are “connected” by virtue of inhibitor binding to the conserved (but thankfully not identical!!) ATP site – this can be leveraged in a family based strategy
-
Strategy
-
-
- Create and share a set of “narrow spectrum” kinase inhibitors that cover the entire screenable kinome
-
Here we briefly describe the latest iteration of our kinase chemogenomic set, progressing toward eventual total kinome coverage. This new edition is called KCGS2.0.
Our kinase chemogenomic set (KCGS) comprises well-annotated inhibitors that target kinases with potent activity but have what we consider narrow-spectrum activity across the kinome. Our goal is to continue growing the set until we have one to three inhibitors for each human kinase. When we reach this point, the set can in principle, be used to determine the relevance and/or function of each kinase in the context of interest. Individually each inhibitor is not promiscuous, and each has defined activity on a narrow set of kinases. When the set is screened in disease-relevant phenotypic assays, one can infer kinase vulnerability based on the results and follow up with more detailed experiments on kinases of interest to confirm the hypothesized dependence.
We have now added additional compounds to KCGS1.0 and created KCGS2.0 affording expanded breadth (more kinases covered) and depth (additional chemotypes for kinase) of coverage. The set is being distributed through cancertools.org which is the research tools arm of Cancer Research UK. Follow this link (https://www.cancertools.org/tools ) and search for KCGS at this tools page.
KCGS2.0 contains 295 compounds and covers 262 kinases. Figure 1 is a plot that depicts the number of citations for each kinase. KCGS2.0 coverage is biased towards more studied (cited) kinases as one might expect, but there are many less well-studied (cited) kinases that are covered by KCGS2.0 (red bars to the right in Figure 1).
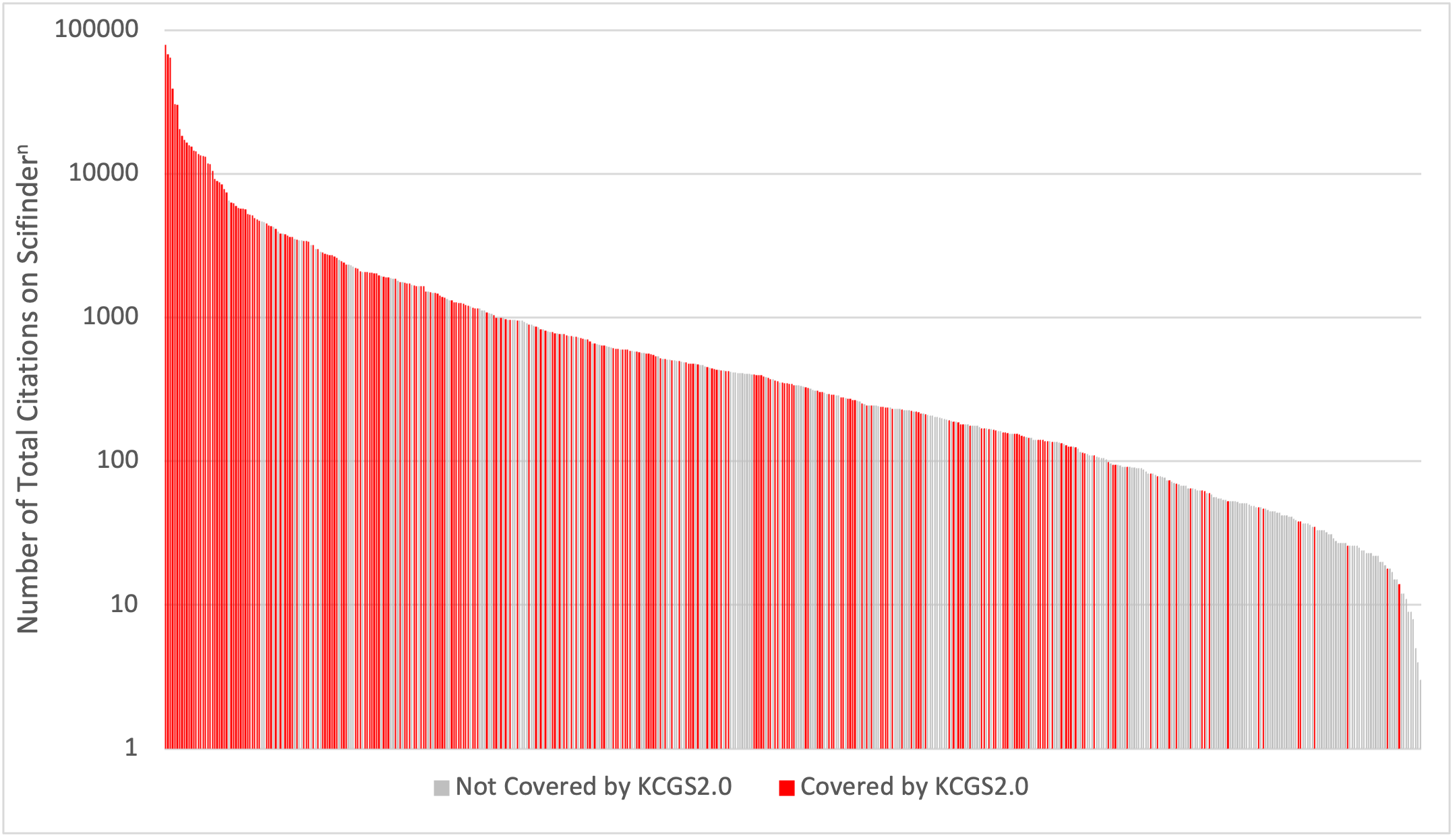
Figure 1: The y-axis (note log scale) provides the number of citations for each kinase. Each bar of the x-axis denotes a compound. The most cited kinases are on the left, and the most understudied kinases are on the far right of the graph. Kinases covered by KCGS2.0 are shown in red bars. Gray bars represent kinases that are not yet covered in our kinase chemogenomic set.
Our kinase chemogenomic set history
The history of both our kinase inhibitor sharing and our kinase chemogenomics work is captured and overviewed here in 5 papers if you’d like to read more. These papers can help you understand why we do this and how our progress has been made possible by open science and collaboration.
- Kinase Open Science
“A public-private partnership to unlock the untargeted kinome” 2013 PMID: 23238671: In this paper we share our goal of illuminating the function of understudied kinases through open science and a public-private partnership.
- The published kinase inhibitor set (PKIS) model
“Seeding collaborations to advance kinase science with the GSK Published Kinase Inhibitor Set (PKIS)” 2014 PMID: 24283969: In this paper we introduce the first set we shared, called PKIS, and the procedure to access the set.
- PKIS
“Comprehensive characterization of the Published Kinase Inhibitor Set” 2016 PMID: 26501955: This paper presents the comprehensive characterization of PKIS compounds.
4. PKIS2
“Progress towards a public chemogenomic set for protein kinases and a call for contributions” 2017 PMID: 28767711: This paper presents the comprehensive characterization of PKIS2 and introduces the concept of building KCGS in continued partnership with the scientific community.
5. KCGS
The Kinase Chemogenomic Set (KCGS): An open science resource for Kinase vulnerability Assessment” 2021: PMID33429995: This paper describes the construction of KCGS and releases the characterization data for KCGS.
6. KCGS2.0
2023: Brand new; you’re seeing it here KCGS2.0 @ Zenodo (https://doi.org/10.5281/zenodo.7855618)!
Frequently Asked Questions
In the summary spreadsheet, what does the S10 (1 mM) mean?
S10 (1 mM) is a selectivity metric generated from Discoverx broad kinome screening data. It is the number of kinases with PoC<10 (equivalent to 90%I) divided by the number of wild-type (non-mutant) kinases screened (generally 403 kinases here). In our case, we screened inhibitors at a concentration of 1 micromolar, thus, the S10 (1 mM). Smaller S10 values represent a more selective compound. Of course, this is an imperfect selectivity measure.
Do you have the same data on all the compounds?
We don’t. Compounds that ended up in KCGS2.0 but started in PKIS may only have data from the Nanosyn panel of assays we ran at that time. In that PKIS experiment, we screened compounds at 100 nM and 1 micromolar. In the spreadsheet of KCGS2.0 summary data, any reference to Nanosyn is talking about the data from the 1 micromolar screening at Nanosyn. Please check out the PKIS paper and supplemental information for more information on the Nanosyn data. Here is the PubMed link: https://pubmed.ncbi.nlm.nih.gov/26501955/
Compounds from PKIS2 that ended up in KCGS2.0 have broad screening data from the Discoverx panel of assays. The workaround KCGS is described here: https://pubmed.ncbi.nlm.nih.gov/33429995/. Please refer to this paper for more detail on the design of KCGS and its use of KCGS. These same guidelines were used in expanding to KCGS2.0, so many of your KCGS2.0 questions may be answered by reading through that paper.
The brand-new compound additions that turn KCGS into KCGS2.0 comes with new, and for the most part unpublished, Discoverx kinome scan data. We have added compounds that cover new kinases (increased breadth of coverage) as well as adding new chemotypes for some kinases (increased depth of coverage).
Are all the compounds exquisitely selective?
Initially, we strived for S10 (1 mM) < 0.03 or so. Many of the compounds only had Nanosyn data initially. We have now tested many of those in the kinomescan assay, and that is reflected in the screening column (KCGS2.0 data overview spreadsheet) if it says “Nanosyn, Discoverx”. These two assay panels are different (but with many overlapping kinases) and have completely different assay formats. In some cases, testing in the Discoverx panel has surfaced additional kinase targets, meaning that compounds with less-than-ideal selectivity are in the set. This just means users of the set need to take this into account as hits from phenotypic screens are followed up.
What are the references you provide in the “reference” column?
When we started building kinase chemogenomic sets and designing new kinase inhibitors, our premise was that we could use kinase inhibitors made for one target as starting points for other kinase targets. The “reference” column provides the original med chem references that report a number of these compounds. If one of these compounds hits in your assay, I encourage you to check out the original paper in case it offers any additional insights. Apologies if we have missed some references. This exercise has demonstrated that useful inhibitors for “other”, often unrelated, kinases can be identified by broad screening of compounds made in medicinal campaigns for another kinase.
If I get a hit from a compound do I know with certainty that the target is critical for my phenotype?
Screening the set will generate hypotheses for you to explore. Any hit in a phenotypic assay needs to be followed up carefully. Of course, looking at the list of targets in row I (target data: generally, Kd<100 nM and/or %I>90 (screened at 1 mM)) is a great place to start. Remember there COULD be other targets. We have not screened all kinases, for example. In addition, S10 (1 mM) is an imperfect selectivity metric. We have highlighted targets with Kd or IC50 < 100 nM, or with >90%I at 1 uM. Targets just slightly weaker than this could also lead to (or contribute to) a phenoytpe. Of course, there may be a chance a compound binds to a nonkinase target. For hits of interest, ALL possibilities should be considered as you seek to link compound to target to mechanism and phenotype.
Acknowledgment
We thank all of our SGC partners, pharma partners, and collaborators around the globe who have helped with this project. We also thank the SGC and the UNC Lineberger Comprehensive Cancer Center for funds that allowed for the resynthesis of KCGS1.0 compounds. We are grateful for funding to our group from the NIH Illuminating the Druggable Genome Project (1U24DK116204-01) and the increased awareness that the IDG program has brought to understudied (and important!) targets.
The SGC is a registered charity (number 1097737) that receives funds from AbbVie, Bayer Pharma AG, Boehringer Ingelheim, Canada Foundation for Innovation, Eshelman Institute for Innovation, Genome Canada, Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD grant no. 115766], Janssen, Merck KGaA Darmstadt Germany, MSD, Novartis Pharma AG, Ontario Ministry of Economic Development and Innovation, Pfizer, Takeda, and Wellcome [106169/ZZ14/Z].