Since we posted our analysis of genetic variation of SARS-CoV-2 main protease (MPro) with inhibitor N3 (PDB: 7bqy), there have been other inhibitors co-crystallized with the MPro (PDB: 6lze and PDB: 6m0k).
MPro acts like a scissor by cutting SARS-CoV-2 polyproteins into functional pieces, making it a critical target for antiviral therapies against SARS-CoV-2.
In June 2020, Dai and colleagues published a paper where they described their structure-based approach to designing two antiviral candidates targeting the MPro of SARS-CoV-2.
They reported two lead compounds, 11a and 11b, and both showed inhibitory activity against the MPro with IC50 values of 53 ± 5 nM and 40 ± 2 nM respectively. Both compounds inhibit MPro through a similar mechanism of action by making covalent linkages via an aldehyde group with cysteine 145 (C145). They also included in vivo pharmacokinetic data for both compounds and concluded that both could be very promising candidates for further development as anti-SARS-CoV-2 candidates.
11a and 11b are pretty much the same molecules with a difference in their p2 region as shown in figure 1 where 11a has a cyclohexyl and 11b has a 3-fluorophenyl at that position.
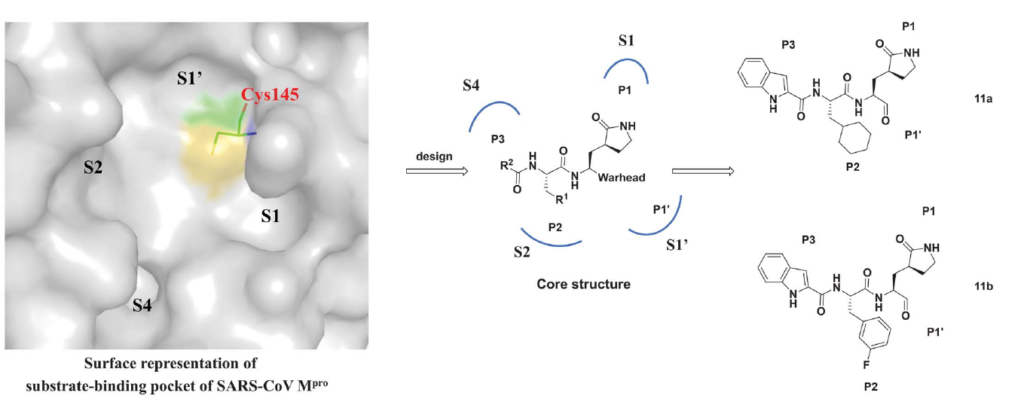
Figure 1. The design strategy for developing inhibitors against SARS-CoV-2 MPro. (Dai et al., 2020)
In this post, I will describe how the non-synonymous mutations identified in more than 15000 SARS-CoV-2 samples at the main protease catalytic site are predicted to affect the binding of these two inhibitors to the MPro.
If you would like to read the details of the previous MPro analysis, please visit this Zenodo report.
According to Nicola De Maio, our collaborator on this project from European Bioinformatics Institute, there were 4 non-synonymous variants at the MPro catalytic site across the SARS-CoV-2 samples from COVID-19 patients. (Table 1)
Next, to predict the effect of those identified mutations on inhibitor (11a and 11b) binding, we use ICM software (Molsoft, SD). In ICM, by mutating the residue, we can see the change in Gibbs free energy of inhibitor binding (ddGbind) which would indicate if the mutation penalizes the binding of the inhibitor significantly or not. Table 1 shows two columns of ddGbind values for each of the inhibitors.
| Residue Number | Wild Type Residue | Mutated Residue | ddGbind (11a) | ddGbind (11b) |
| 49 | Met | Ile | 4.8 | 0.4 |
| 52 | Pro | Ser | -0.1 | -0.1 |
| 142 | Asn | Ser | 1.3 | 0.7 |
| 168 | Pro | Ser | 0.7 | 0.7 |
Table 1. The predicted change in Gibbs free energy associated with the non-synonymous variants found at MPro catalytic site across more than 15000 SARS-CoV-2 samples. ddGbind values are in Kcal/mol.
While M49I is predicted to penalize 11a binding to MPro significantly (ddGbind > 2.0 Kcal/mol), that is not the case with 11b. M49 is juxtaposed to the functional group that is diverging between the two inhibitors (cyclohexyl in 11a, 3-fluorophenyl in 11b), which could be a reason for the observed difference (Fig. 2). However, we also note a small but non-negligible variation backbone conformation of the loop where M49 is located (Fig. 2). Importantly, sidechains are flexible, but the protein backbone is not when calculating ddGbind with ICM. It is, therefore, possible that an Ile is acceptable at position 49 in the context of the loop conformation observed in one structure and not the other. Molecular dynamics simulations may help better analyze the effect of the M149I mutation on the binding 11a. At the moment we cannot reliably predict this effect.
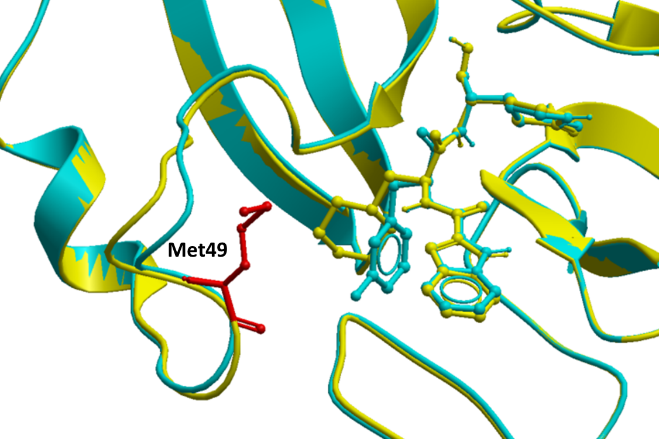
Figure 2. M49 in proximity to 11a (PDB: 6lze – yellow) and 11b (PDB: 6m0k – cyan) in the SARS-CoV-2 MPro crystal structure.
Lastly, we used two-dimensional diagrams as shown in figure 3 to illustrate the chemical interactions between 11a and 11b and how they are different from one another.
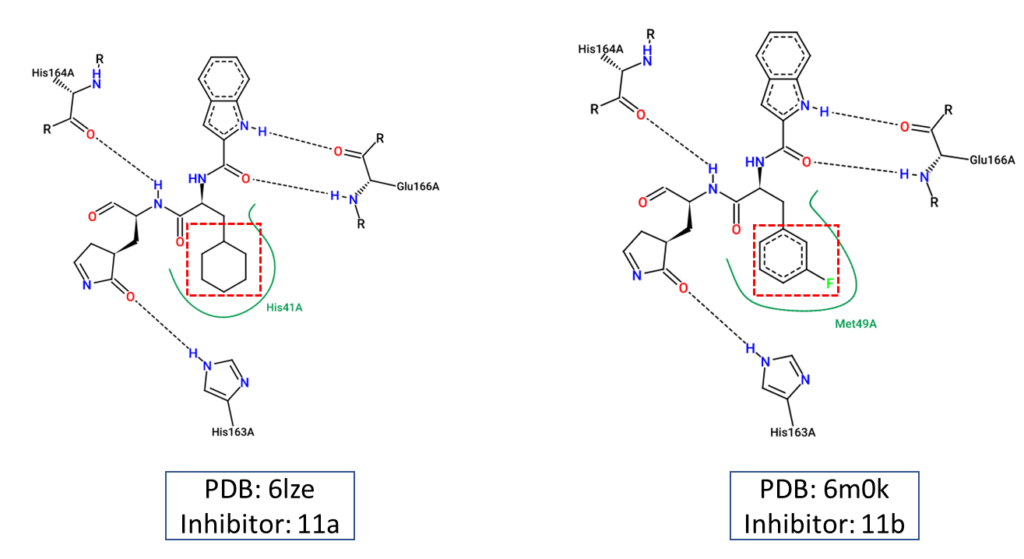
Figure 3. Two-dimensional diagrams representing the chemical interactions between the main protease and the two inhibitors, 11a and 11b. These diagrams were created using Pose.View in Proteins.Plus.
From the ddGbind values in table 1, we can conclude that the variants found in SARS-CoV-2 samples are both rare and only expected to minimally affect the binding of 11b to MPro.
We created an energy matrix where we consider the effect of every possible mutation at the MPro catalytic site on the binding of 11a and 11b with MPro. Table 2 includes a list of 20 residues lining the MPro catalytic pocket. No mutations at Cys145 were considered since it makes the covalent linkage with the inhibitors 11a and 11b which is essential for their inhibitory mechanisms against MPro.
It is likely that the residues of the rows with green color are not making optimal interactions with the inhibitors. This is reflected in the very slight and non-significant lowering in ddGbind values of these residues (mutations are stabilizing) and hence the green color. On the other hand, the sidechains whose mutations are shown in orange are likely the positions where the MPro would better interact with its inhibitors. As a result, the mutations of those sidechains are predicted to penalize inhibitor binding significantly and hence the orange color as shown in Table 2 and Table 3.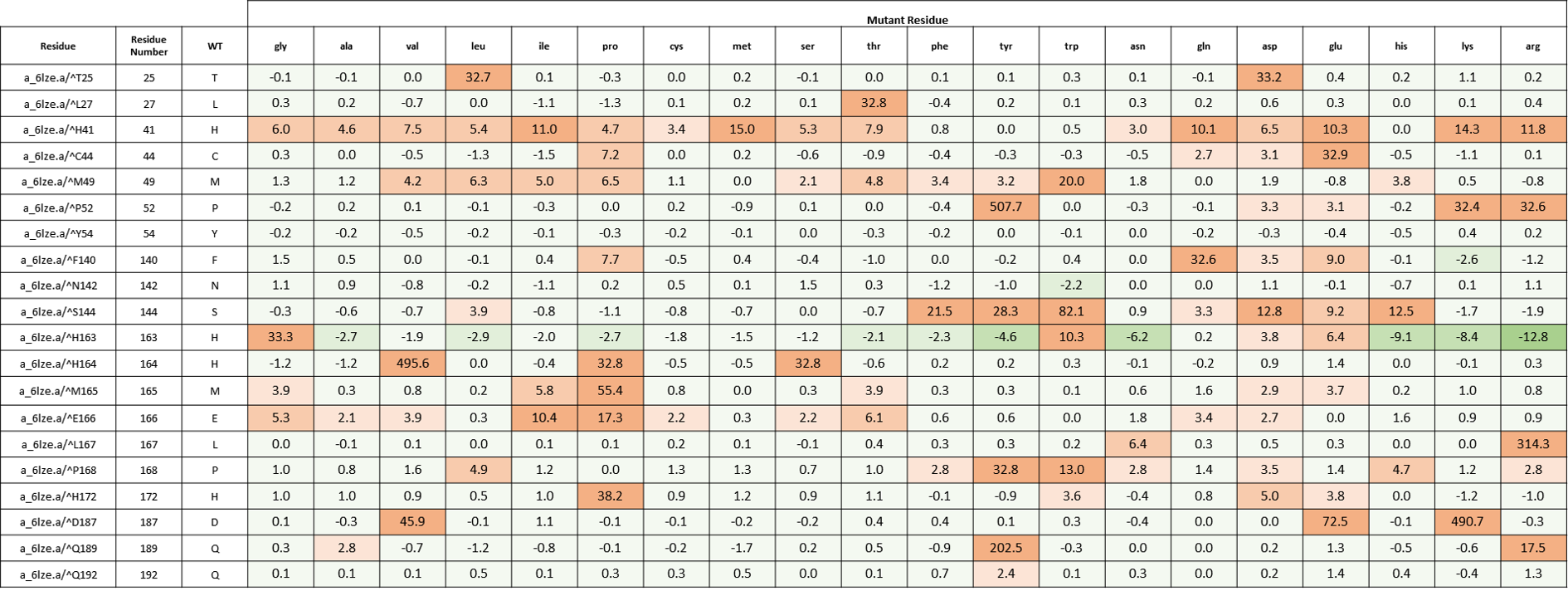 Table 2. The energy matrix showing the effect of all possible mutations at the SARS-COV-2 MPro catalytic site and the predicted effect on 11a binding (PDB: 6lze). The energy unit is kcal/mol. WT = wild type residue.
Table 2. The energy matrix showing the effect of all possible mutations at the SARS-COV-2 MPro catalytic site and the predicted effect on 11a binding (PDB: 6lze). The energy unit is kcal/mol. WT = wild type residue.

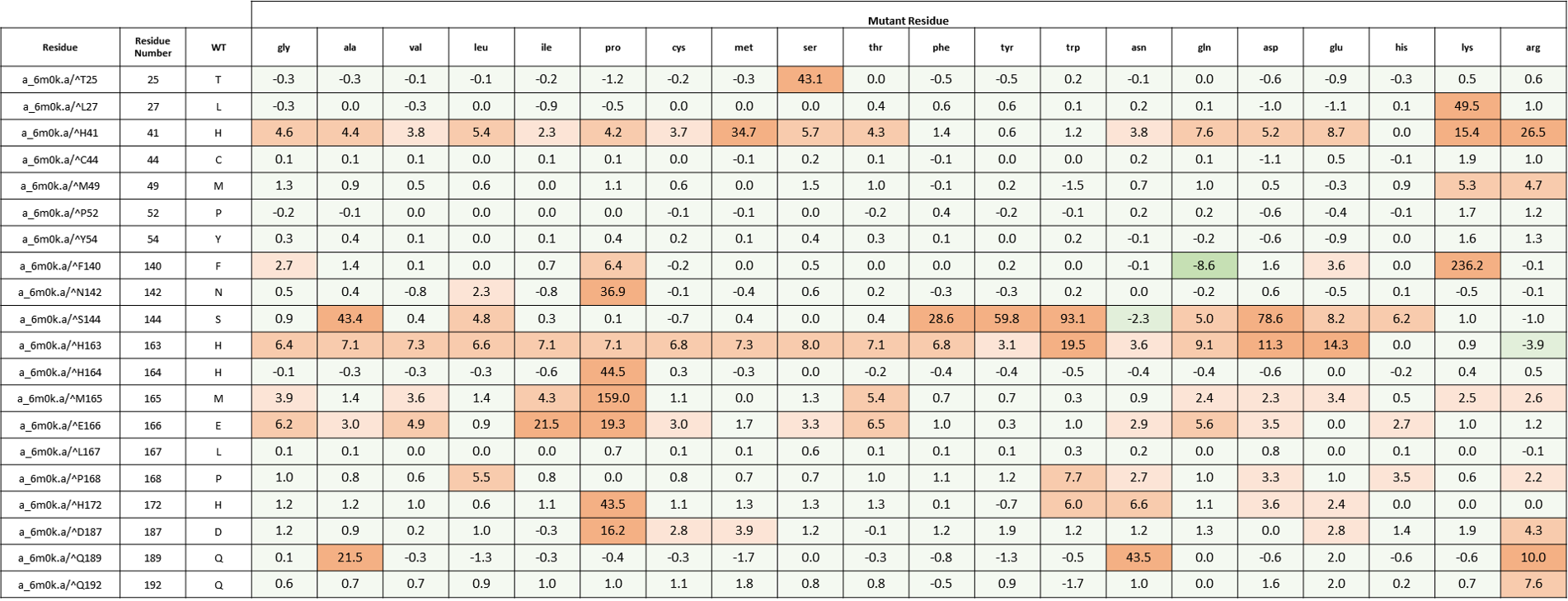 Table 3. The energy matrix showing the effect of all possible mutations at the SARS-COV-2 MPro catalytic site and the predicted effect on 11b binding (PDB: 6m0k). The energy unit is kcal/mol. WT = wild type residue.
Table 3. The energy matrix showing the effect of all possible mutations at the SARS-COV-2 MPro catalytic site and the predicted effect on 11b binding (PDB: 6m0k). The energy unit is kcal/mol. WT = wild type residue.
Please contact me via the “Leave a comment” link at the top of this post. Stay Tuned for more updates on this project.
References:
Dai, W., Zhang, B., Jiang, X., Su, H., Li, J., Zhao, Y., Xie, X., Jin, Z., Peng, J., Liu, F., Li, C., Li, Y., Bai, F., Wang, H., Chen, X., Cen, X., Hu, S., Yang, X., Wang, J., … Liu, H. (2020). Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science, 368(6497), 1331-1335. https://doi.org/10.1126/science.abb4489