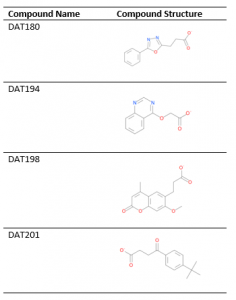
Based on previous 19F NMR and SPR assay experiments, 4 compounds were used to set up co-crystallization experiments (Table 1). I was able to get co-crystal structures of 3 of the 4 compounds! You can see full experimental details on Zenodo.
Table 1. USP5 Zf-UBD ligands
All the compounds occupied the ubiquitin binding pocket, which is in agreement with the 19F NMR data. The electron density of the compounds were quite clear in the binding pocket of USP5 Zf-UBD. An omit map of DAT180 is shown in Figure 1.
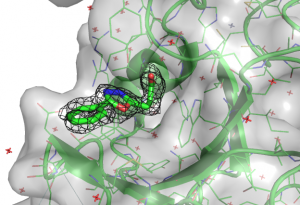
Figure 1. Omit map (σ2) of USP5171-290 and DAT180
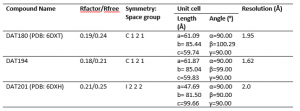
It is to be noted that DAT180 and DAT201 structures are now refined, were deposited in the PDB (6DXT, 6DXH) and will be available from there soon. Table 2 summarizes the solved co-crystal structures for each compound.
Table 2. Summary of co-crystal structures
Interestingly, a loop in the vicinity of DAT201 adopted a conformation not seen in previous structures of USP5 (Figure 2). The novel cavity resulting from this conformational shift may be exploited for the design of follow-up compounds.
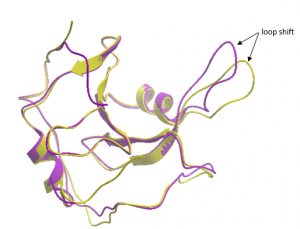
Figure 2. Overlay of DAT201 structure (yellow) & PDB: 2G43 (purple)
The solved crystal structure of DAT194 was the most interesting of the solved structures. We saw electron density in the binding pocket but it did not correlate with the structure of the compound we had ordered! After some discussion, we tried to see if modeling an isomer of that compound would “fit” and it fit beautifully (Figure 3b). Both DAT194 and the Figure 3b isomer have the same mass. The purity of DAT194 was >95% when checked by UPLC/MS. It’s very cool to see that the high resolution crystal structure can differentiate between isomers.
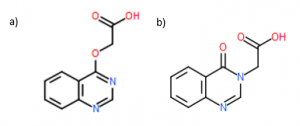
Figure 3. a) Expected DAT194 structure b) Modeled isomer with same mass as DAT194
In the future, I’ll be using computational techniques to prioritize chemical analogs that may better exploit the binding pocket. Decorating the core scaffold of the compounds with different chemical groups can potentially increase ligand affinity to the protein domain. Stay tuned!
2 Replies to “Co-crystal structures of USP5 Zf-UBD and weak binding compounds”