The RNA-dependent RNA-polymerase (RdRp) also known as non-structural protein 12 (nsp12) is the target of antiviral agent remdesivir. Nsp12 has an important role in viral genome replication and transcription. (Chen et al., 2020)
Chen et al. identified a new pocket on the N-terminal extension of nsp12 occupied by ADP-Mg2+ after solving the structure of the helicase-polymerase complex. This ADP-bound pocket is on the N-terminal nidovirus RdRp-associated nucleotidyltransferase (NiRAN) domain which is also well-resolved. (Chen et al., 2020).
Based on conservation profile across nidovirales (an order of positive-stranded RNA viruses including SARS-CoV-2), the NiRAN domain is believed to have nucleotidylation activity, though the protein substrates to which nucleotides are added by the NiRAN domain are unknown. Reverse genetics was used to establish the importance of NiRAN domain for nidoviral replication: mutating residues conserved in NirAN domains induces significant reduction of SARS-CoV-1 and Equine viral arteritis (EAV) viral titers in cell culture (Lehmann et al., 2015). Previous structural homology search of the SARS-CoV-1 nsp12 NiRAN domain showed significant homology with protein kinases, including some key catalytic residues such as Asn209 and NiRAN conserved residues As218 and Phe219. (Kirchdoerfer et al., 2019)
For our project, we were interested in assessing the druggability of this pocket and finding out how conserved is this site both across past and novel coronaviruses and across SARS-CoV-2 samples from COVID-19 patients.
To achieve our first aim, we used a tool called sitemap (Schrodinger, NY) to assess the druggability of the ADP-bound pocket of NiRAN domain of RdRp using the available nsp12 structure in the protein data bank (PDB code: 6xez). The result indicates that this highly polar pocket is a difficult druggable site (Dscore = 0.85) as shown in figure 1. (Halgren, 2009)
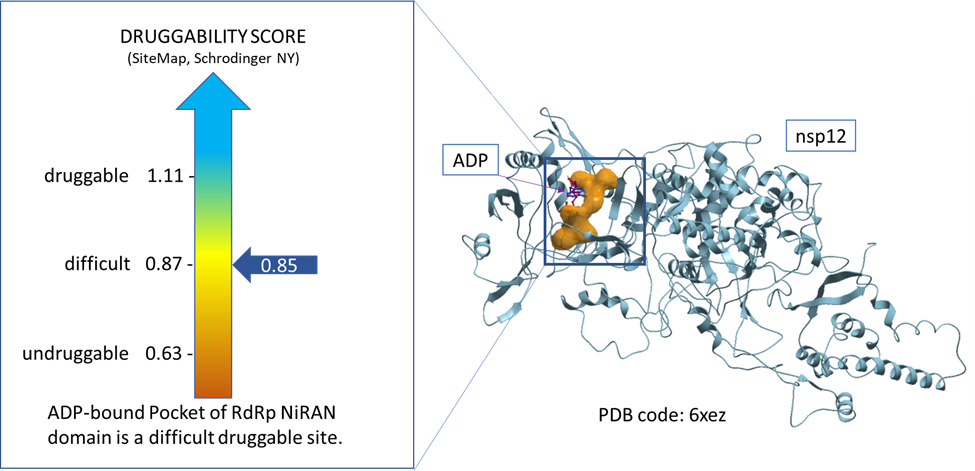
Figure 1. Druggability assessment of the ADP-bound pocket of NiRAN domain of RdRp.
We picked 30 amino acid residues that line this site of interest as identified by their proximity (~2.8Å) to both the bound ADP molecule and the Van der Waals boundaries of the corresponding pocket mapped by ICM. (Molsoft, San Diego). (Figure 2, Zenodo report)
As part of our second aim, we conducted a broad survey of viral proteins from Alpha- and Betacoronaviruses to identify the most conserved druggable sites. By integrating variability and druggability data we can infer the most promising strategies for developing broad-spectrum viral inhibitors against coronaviruses. This analysis can be valuable in the context of the emergence of future coronaviruses circulating in bat reservoir species.
To assess this site’s genetic variability, we made a diversity dendrogram by performing a multiple sequence alignment using reviewed sequences from Uniprot, of which 6 are from the Alphacoronavirus genus and 21 from the Betacoronavirus genus. (Figure 3, Zenodo report) We observe that 21 out of 30 residues are identical at this site of RdRp which translates into 70% identity.
As a follow-up to assessing the genetic variability among Alphacoronavirus and Betacoronavirus entries, we also looked at the conservation of this site across SARS-CoV-2 samples. Our collaborator, Nicola De Maio, from Nick Goldman’s lab at the European Bioinformatics Institute has identified the non-synonymous mutations at ADP-bound pocket of NiRAN domain of RdRp across more than 39000 SARS-CoV-2 samples. Altogether he identified non-synonymous mutations at 6 unique amino acid residues consisting of 6 unique non-synonymous variants. In table 1 of the Zenodo report, there are details about the mutation events underlying these variants.
Comparison with previously analyzed RdRp pockets, RdRp active site, and allosteric site (M666 pocket):
We previously analyzed the druggability and conservation of the catalytic site and an ectopic site (that we called M666 pocket) of RdRp. We find that the and ectopic pocket (Dscore = 1.03) is more druggable than the catalytic pocket (Dscore 0.86) occupied by RNA-conjugated Rmedesivir and NiRAN pocket (Dscore=0.85).
On the other end, the catalytic site of RdRp is among the most conserved pockets that we have seen across SARS-CoV-2 protein structures (83% conserved in coronaviruses) followed by the NiRAN pocket (68% conserved) and the M666 pocket (33% conserved).
We observe a related trend when looking at mutation rates across COVID-19 samples: the catalytic site and NiRAN pocket were genetically more stable, with 6 unique mutations found in 6 samples each, while the M666 pocket was more prone to mutations (10 mutations found in 13 variants).
A strategy may be to develop compounds that include a chelating moiety to engage the Mg2+ observed in the ADP-bound NiRAN pocket. This site has reasonable genetic stability and is potentially an interesting target for the development of broad-spectrum inhibitors.
You can view this report on Zenodo as well.
Please contact me via the “Leave a comment” link at the top of this post. Stay Tuned for more updates on this project.
References
Chen, J., Malone, B., Llewellyn, E., Grasso, M., Shelton, P. M., Olinares, P. D., Maruthi, K., Eng, E., Vatandaslar, H., Chait, B. T., Kapoor, T., Darst, S. A., & Campbell, E. A. (2020). Structural basis for helicase-polymerase coupling in the SARS-Cov-2 replication-transcription complex. https://doi.org/10.1101/2020.07.08.194084
Halgren, T. A. (2009). Identifying and characterizing binding sites and assessing Druggability. Journal of Chemical Information and Modeling, 49(2), 377-389. https://doi.org/10.1021/ci800324m
Kirchdoerfer, R. N., & Ward, A. B. (2019). Structure of the SARS-Cov NSP12 polymerase bound to NSP7 and NSP8 Co-factors. https://doi.org/10.1101/551986
Lehmann, K. C., Gulyaeva, A., Zevenhoven-Dobbe, J. C., Janssen, G., Ruben, M., Overkleeft, H. S., Van Veelen, P. A., Samborskiy, D., Kravchenko, A. A., Leontovich, A. M., Sidorov, I. A., Snijder, E. J., Posthuma, C. C., & Gorbalenya, A. E. (2015). undefined. Nucleic Acids Research, 43(17), 8416-8434. https://doi.org/10.1093/nar/gkv838