Joel K. Annor-Gyamfi, Felix Nwogbo, Kun Qian, Yuhong Du, Kenneth H. Pearce Jr, Opher Gileadi, Haian Fu, Stephen V. Frye and Alison D. Axtman
https://doi.org/10.5281/zenodo.4685122
Alzheimer’s disease (AD) is the most common cause of dementia worldwide but very few therapeutic options exist despite multiple high-profile but failed clinical trials. And as the population of the United States ages, AD and cognitive failure will become an increasing problem that will have a huge impact on society and the economy. As a result, the National Institute on Aging (NIA) has created and funds the TREAT-AD (TaRget Enablement to Accelerate Therapy for AD) program. One of the goals of the TREAT-AD is to create small molecules as high-quality chemical probes to modulate unexplored protein targets implicated in AD. It is hoped that these probes will help to advance understanding of the pharmacological intervention points most likely to lead to new therapies for the treatment of this affliction.
In support of this purpose, the moesin (MSN)-CD44 pathway has been identified by the group lead, Allan Levey at Emory during the Accelerating Medicines Partnership-Alzheimer’s Disease (AMP-AD) phase of the TREAT-AD program, as a potential target for therapeutic intervention in AD. Moesin, a cytoskeletal protein with roles in focal adhesion-mediated cell motility, has been implicated in AD pathology due to the presence of MSN-positive microglia-like cells around amyloid plaques and tangle-like structures in AD brains. Both MSN and CD44 have emerged as key inflammation hub proteins in an investigation of the AD brain proteome, along with weighted co-expression network analysis.1 Moreover, an increased CD44 gene expression in lymphocytes derived from AD patients has been reported.2 Thus, we hypothesize that CD44 antagonism would reduce AD progression via dampening microglia activation and reducing infiltration of circulating immune cells.
Katis et al., 2020 also used co-crystal structures of moesin and the related FERM protein EPB41L3 to demonstrate how the intracellular tail of CD44 (a transmembrane receptor) binds to MSN. This binding mode guided the development of Homogeneous Time-Resolved Fluorescence (HTRF)-based assays for high-throughput screening (HTS) by the Gileadi lab at Oxford University for molecules that disrupt the CD44-MSN interaction.1 An initial ultra-HTS at Emory University by the Fu lab resulted in 231 primary hits, out of which 120 compounds repeated in dose-response. Subsequently, a pull-down assay using full-length protein confirmed interruption of the protein-protein interaction by three fused tetrahydroquinoline (THQ) analogues (Figure 1).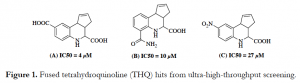
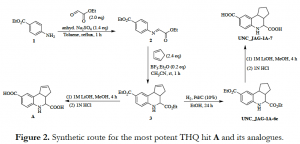
A robust synthetic plan was outlined to remake these hits for further testing. A representative scheme for the synthesis of the most potent hit A is shown below in Figure 2. Hits B and C can be accessed via a similar route when their respective aniline derivatives are used. The outlined route involves the formation of a Schiff base 2 from the condensation reaction between ethyl 4-aminobenzoate (1) and ethyl glyoxalate under reflux conditions. 2 undergoes a [4+2] Diels-Alder reaction with a freshly distilled cyclopentadiene in the presence of boron trifluoride etherate to give 3, which is further subjected to ester hydrolysis with 1M lithium hydroxide in methanol, followed by 1N HCl to afford hit A. LC-MS confirmed the masses of the intermediates and products.
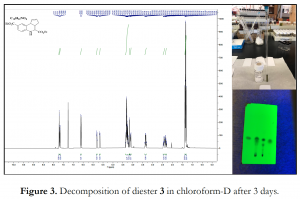
Additionally, 1H Nmr data of the diacid A and diester 3 were obtained in DMSO-d6 and CDCl3 respectively to confirm their identity and purity. However, discoloration of the clear solution in the Nmr tubes was observed after 3 days at room temperature – a similar observation was made when A was sent for testing in a biochemical assay. Although 1H Nmr analysis of the discolored sample of 3 did not differ from the pure form, TLC in 20% ethyl acetate/hexane eluent revealed significant decomposition in solution over time (Figure 3). TLC of the diacid A is not shown because it was hard to see any separation due to streaking/tailing on the TLC plate. Moreover, a few milligrams of hits A and B were outsourced from commercial vendors and they both demonstrated similar decomposition traits in solution.
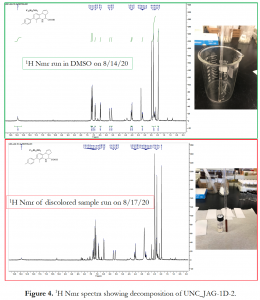
A similar observation was made for the monoacid THQ analogue, UNC_JAG-1D-2. For this compound, the 1H Nmr spectrum of the decomposed product was significantly different from the pure sample, both in the aromatic and aliphatic regions of the spectrum (Figure 4).
The preparation of the monoacid as shown in Figure 5, started with esterification of the commercially available chloro-substituted THQ acid (Chembridge ID: 5659221) to give ester 4. Suzuki coupling of 4 with p-tolylboronic acid in the presence of Pd2(dba)3 and Xphos as catalyst and ligand, respectively, affords intermediate 5. Compound 5 was subjected to alkaline hydrolysis with 1M lithium hydroxide in THF, followed by treatment with 1N HCl to yield UNC_JAG-1D-2. An initial attempt to convert the commercial chloro-substituted THQ acid (Chembridge ID: 5659221) to the coupled acid product, UNC_JAG-1D-2, via a single step approach was unsuccessful.
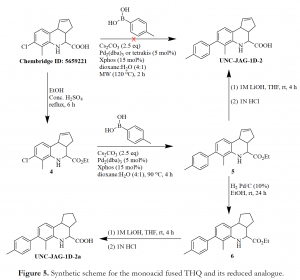
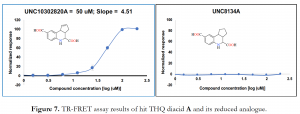
In our quest to ascertain which aspect of these tricyclic systems was responsible for decomposition, we made the following structural modifications. First, we reduced the double bond in the cyclopentene ring of hit diacid A, diester intermediate 3 and monoacid UNC_JAG-1D-2 to afford their respective saturated analogues; UNC_JAG-1A-7, UNC_JAG-1A-6e (shown in Figure 2), and UNC_JAG-1D-2a (Figure 5). Moreover, we also analyzed THQ as well as dihydroquinoline analogues which did not have the fused cyclopentene ring (Figure 6). Consequently, the integrity of these structurally modified analogues in solution was maintained, as no discoloration was observed even after 5 days of sitting. However, this effort came with a cost of losing the activity of these analogues in the Time-Resolved Fluorescence Resonance Energy Transfer (TR-FRET) assay run by the TREAT-AD Medicinal Chemistry core at UNC. Most importantly, the hit diacid A which had demonstrated an initial activity in the assay was completely inactive when its double bond was removed (Figure 7) – an indication that the double bond in the cyclopentene ring was at least partially responsible for the observed activity, and decomposition.
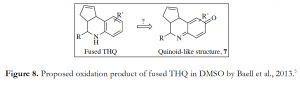
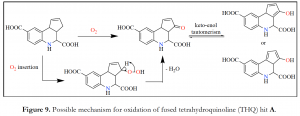
As we continued our search for explanations of why the double bond could potentially be a liability in the fused THQ, we chanced upon two articles from Geiss et al., 2011 and Suape et al., 2011 which describe these scaffolds as frequent screening hits in several bioassays.3,4 Thus, the question of whether these fused THQ compounds were privileged or promiscuous was left to be answered. A more recent report from Baell et al., 2013 classified these scaffolds as Pan Assay Interference compouNdS (PAINS). While these compounds appeared to be selective, optimizable and valid, they were found to be non-progressable often after a significant application of resources. The authors proposed that these fused THQs could be oxidized in DMSO screening samples to form traces of quinoid-like structure 7 (Figure 8), which could potentially interfere in a bioassay to give a false positive outcome.5 However, based on our own observations, we believe that the double bond in the cyclopentene ring makes the fused THQ analogues susceptible to oxidative decomposition in solution to form traces of compounds that could interfere in our assay to give a false positive result. Thus, we propose a possible mechanism shown in Figure 9 for such oxidation involving the double bond under consideration. Although we are no longer moving forward with these fused THQ analogues due to their structural liability, efforts are ongoing to identify and isolate the decomposition product(s).
Chemistry Experimental
Ethyl (E/Z)-4-((2-ethoxy-2-oxoethylidene)amino)benzoate (2): To a 10 mL round-bottom flask equipped with a reflux condenser was added ethyl 4-aminobenzoate (0.36 g, 2.15 mmol), ethyl glyoxalate (50% solution in toluene, 426 µL, 4.30 mmol) and sodium sulfate (0.46 g, 3.23 mmol) in toluene (3 mL), and the reaction was heated to 110 oC for 30 min. The reaction was cooled, filtered and the solvent was concentrated in vacuo. The oily residue was used in the next step without further purification.
Diethyl 3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4,8-dicarboxylate (3): Compound 2 (1.50 g, 6.02 mmol) and acetonitrile (2.5 mL) were charged to a 50 mL round-bottom flask. The flask was fitted with a spectrum and purged with nitrogen balloon. Boron trifluoride etherate (149 µL, 1.20 mmol) was added via syringe, followed by the addition of freshly distilled cyclopentadiene (988 µL, 12.04 mmol) and the reaction was stirred at room temperature for 2 h. The reaction mixture was diluted with H2O and extracted with EtOAc (3 x 15 mL). The combined organic layers were washed with brine, dried with Na2SO4 and the solvent was concentrated in vacuo. The crude material was purified by column chromatography (silica, 0-100% EtOAc in hexane) to afford 3 as an off-white solid (950 mg, 50%). 1H NMR (400 MHz, CDCl3): δ 7.73 – 7.69 (m, 1H), 7.67 (dd, J = 8.3, 2.0 Hz, 1H), 6.61 (d, J = 8.4 Hz, 1H), 5.81 (ddt, J = 5.8, 2.9, 1.5 Hz, 1H), 5.66 (dd, J = 5.8, 2.4 Hz, 1H), 4.38 – 4.19 (m, 5H), 4.16 (d, J = 3.6 Hz, 1H), 4.12 – 4.08 (m, 1H), 3.36 (tt, J = 9.0, 4.4 Hz, 1H), 2.48 – 2.40 (m, 1H), 2.33 (ddt, J = 11.9, 9.0, 3.4 Hz, 1H), 1.37 – 1.34 (m, 3H), 1.34 – 1.31 (m, 3H). LCMS: expected mass for [M+H]+ (C18H22NO4): 316.15, found: 316.
Ethyl 7-chloro-6-methyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylate (4): To a 150 mL round-bottom flask equipped with a reflux condenser was added 7-chloro-6-methyl-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylic acid (2.64 g, 10.0 mmol) and concentrated sulfuric acid (375 µL, 7.0 mmol) in ethanol (50 mL), and the reaction mixture was heated at 78 oC for 6 h. The reaction mixture was diluted with H2O and extracted with EtOAc (3 x 25 mL). The combined organic layers were washed with brine, dried with Na2SO4 and the solvent was concentrated in vacuo to yield 4 as a colorless oil (2.80 g, 95%). 1H NMR (400 MHz, CDCl3) δ 6.81 (d, J = 8.2 Hz, 1H), 6.76 (d, J = 8.2 Hz, 1H), 5.71 – 5.67 (m, 1H), 5.66 – 5.61 (m, 1H), 4.38 – 4.21 (m, 2H), 4.07 (dd, J = 7.3, 2.9 Hz, 2H), 3.34 (qd, J = 8.9, 3.5 Hz, 1H), 2.45 (ddq, J = 15.7, 8.4, 2.4 Hz, 1H), 2.33 (ddt, J = 16.2, 9.0, 2.2 Hz, 1H), 2.23 (s, 3H), 1.33 (t, J = 7.1 Hz, 3H). LCMS: expected mass for [M+H]+ (C16H19ClNO2): 292.10, found: 292.
Ethyl 6-methyl-7-(p-tolyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylate (5): To a microwave vial were added 4 (210.8 mg, 722.5 µmol), p-tolylboronic acid (147.3 mg, 1.08 mmol), Pd2(dba)3 (33.1 mg, 36.12 µmol), dicyclohexyl(2′,4′,6′-triisopropyl-[1,1′-biphenyl]-2-yl)phosphane (51.7 mg, 108.4 µmol) and Cs2CO3 (494.3 mg, 1.52 mmol) in dioxane:H2O (v/v, 4:1, 7.5 mL). The vial was fitted with a spectrum and purged with nitrogen balloon. The reaction vessel was sealed and heated to 90 oC for 4 h. The reaction was cooled, filtered through celite and the solvent was concentrated in vacuo. The product was used in the next step without further purification.
General procedure for saponification: To a 50 mL round-bottom flask was suspended the tetrahydroquinoline ester derivative (3, 5, 6, UNC_JAG-1A-6e; 50 mg) in 1M LiOH (5 mL). THF or MeOH was added until a clear solution remained. The reaction mixture was stirred for 4 h at room temperature and then neutralized with 1N HCl (10 mL). Extraction with EtOAc (3 x 20 mL) gave the combined organic layers which were washed with brine, dried with Na2SO4 and concentrated in vacuo to yield the desired product.
3a,4,5,9b-Tetrahydro-3H-cyclopenta[c]quinoline-4,8-dicarboxylic acid (A): Compound 3 (50 mg, 0.16 mmol) was dissolved in MeOH. Yield: Light brown solid (39 mg, 95%); 1H NMR (400 MHz, DMSO-d6) δ 12.51 (s, 2H), 7.52 (s, 1H), 7.43 (dd, J = 8.4, 2.1 Hz, 1H), 6.79 (d, J = 8.4 Hz, 1H), 6.09 (s, 1H), 5.83 – 5.78 (m, 1H), 5.61 (d, J = 6.7 Hz, 1H), 4.06 (d, J = 3.3 Hz, 1H), 4.01 (d, J = 8.3 Hz, 1H), 3.17 – 3.09 (m, 1H), 2.33 – 2.16 (m, 2H). LCMS: expected mass for [M+H]+ (C14H14NO4): 260.08, found: 260.
2,3,3a,4,5,9b-Hexahydro-1H-cyclopenta[c]quinoline-4,8-dicarboxylic acid (UNC_JAG-1A-7): Compound UNC_JAG-1A-6e (50 mg, 0.16 mmol) was dissolved in MeOH. Yield: Tan solid (34 mg, 82%); H NMR (400 MHz, DMSO-d6) δ 12.43 (s, 2H), 7.57 (s, 1H), 7.43 (dd, J = 8.4, 2.0 Hz, 1H), 6.72 (d, J = 8.4 Hz, 1H), 6.23 (s, 1H), 4.04 (d, J = 3.4 Hz, 1H), 2.67 – 2.59 (m, 1H), 2.03 (dq, J = 15.0, 7.9 Hz, 1H), 1.86 – 1.21 (m, 6H). LCMS: expected mass for [M+H]+ (C14H16NO4): 262.10, found: 262.
6-Methyl-7-(p-tolyl)-3a,4,5,9b-tetrahydro-3H-cyclopenta[c]quinoline-4-carboxylic acid (UNC_JAG-1D-2): Compound 5 (50 mg, 0.14 mmol) was dissolved in THF. Yield: Tan solid (43 mg, 93%); 1H NMR (400 MHz, DMSO-d6) δ 12.94 (s, 1H), 7.19 (d, J = 7.7 Hz, 2H), 7.11 (d, J = 7.7 Hz, 2H), 6.89 (d, J = 7.8 Hz, 1H), 6.48 (d, J = 7.7 Hz, 1H), 5.76 – 5.71 (m, 1H), 5.59 (d, J = 5.6 Hz, 1H), 4.06 (d, J = 9.5 Hz, 1H), 4.01 (d, J = 3.5 Hz, 1H), 3.18 (dt, J = 11.5, 5.7 Hz, 1H), 2.32 (s, 6H), 1.96 (s, 3H). LCMS: expected mass for [M+H]+ (C21H22NO2): 320.16, found: 320.
6-Methyl-7-(p-tolyl)-2,3,3a,4,5,9b-hexahydro-1H-cyclopenta[c]quinoline-4-carboxylic acid (UNC_JAG-1D-2a): Compound 6 (50 mg, 0.14 mmol) was dissolved in THF. Yield: Tan solid (44 mg, 95%); 1H NMR (400 MHz, Methanol-d4) δ 7.16 (d, J = 7.8 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 6.90 (d, J = 7.9 Hz, 1H), 6.47 (d, J = 7.9 Hz, 1H), 3.80 (d, J = 3.3 Hz, 1H), 3.41 – 3.34 (m, 1H), 2.82 (dt, J = 10.7, 5.3 Hz, 1H), 2.35 (s, 3H), 2.12 – 2.05 (m, 1H), 2.01 (s, 3H), 1.78 – 1.46 (m, 6H). LCMS: expected mass for [M+H]+ (C21H24NO2): 322.17, found: 322.
General procedure for hydrogenation: To a 10 mL round-bottom flask was added the tetrahydroquinoline ester derivative (UNC_JAG-1A-5e, 5; 50 mg) and ethanol (2.5 mL). The flask was fitted with a spectrum and purged with nitrogen balloon. Palladium on carbon (10% w/w, 5 mg, 0.02 mmol) was added and the mixture was run under hydrogen at room temperature for 24 h. Filtration of the dark solid through celite gave a liquid residue which was concentrated in vacuo to afford the desired reduced product.
Diethyl 2,3,3a,4,5,9b-hexahydro-1H-cyclopenta[c]quinoline-4,8-dicarboxylate (UNC_JAG-1A-6e): derived from compound UNC_JAG-1A-5e. Yield: Off-white solid (49 mg, 98%); 1H NMR (400 MHz, CDCl3) δ 7.79 (s, 1H), 7.66 (d, J = 8.4 Hz, 1H), 6.54 (d, J = 8.4 Hz, 1H), 4.69 (s, 1H), 4.36 – 4.18 (m, 5H), 4.13 (d, J = 3.4 Hz, 1H), 3.39 (s, 1H), 2.81 (d, J = 9.0 Hz, 1H), 2.13 – 2.04 (m, 1H), 1.92 (s, 1H), 1.53 – 1.44 (m, 3H), 1.33 (dt, J = 14.1, 7.1 Hz, 6H). LCMS: expected mass for [M+H]+ (C18H24NO4): 317.16, found: 318.
Acknowledgement
This work was supported by the NIH National Institutes on Aging TREAT-AD Center for Alzheimer’s Disease grant U54 AG065187.
References
- Katis, V.; Bradshaw, W.; Betarbet, R.; Jimenez-Antunez, C.; Seyfried, N.T.; Slakman, B.; Mulhern, C.; Guie, M-A.; Keefe, A.; Guilinger, J.; Zhang, Y.; Huguet, C.; Levey, A.I.; Brennan, P.E.; Mangravite, L.M.; Gileadi, O.; The Open-AD Consortium. Targeting the Moesin-CD44 pathway for therapeutic intervention in Alzheimer’s disease. Alzheimer’s Dement. 2020, 16 (Suppl. 3),
- Uberti, D.; Cenini, G.; Bonini, S.A.; Barcikowska, M.; Styczynska, M.; Szybinska, A.; Memo, M. Increased CD44 gene expression in lymphocytes derived from Alzheimer disease patients. Neurodegenerative Dis. 2010, 7, 143-147.
- Geiss, B.J.; Stahla-Beek, H.J.; Hannah, A.M.; Gari, H.H.; Henderson, B.R.; Saeedi, B.; Keenan, S.M. A high-throughput screening assay for the identification of flavivirus NS5 capping enzyme GTP-binding inhibitors: Implications for antiviral drug development. Biomol. Screen. 2011, 16, 852-861.
- Saupe, J.; Roske, Y.; Schillinger, C.; Kandem, N.; Radetzki, S.; Diehl, A.; Oschnkinat, H.; Krause, G.; Heinemann, U.; Rademann, J. Discovery, structure-activity relationship studies, and crystal structure of nonpeptide inbitors bound to the Shank3 PDZ domain. ChemMedChem 2011, 6, 1411-1422.
- Baell, J.B.; Ferrins, L.; Falk, H.; Nikolakopoulos, G. PAINS: Relevance to tool compound discovery and fragment-based screening. Aust. J. Chem. 2013, 66, 1483-1494.