In this post, I would like to share my recent work on HAO1 fragment screening by crystallography, as a means to identify inhibitor starting points.
So why fragment screening by x-ray crystallography? Well there are two components to my method of choice:
Fragments instead of complex compounds provide good starting chemistry
Fragments are less than 300 Daltons in size, in contrast to the larger, more complex compounds conventionally used in high throughput screening. This review summarizes the merits of using fragments for drug discovery, which include:
- A few hundred fragments explore the same diversity of chemical groups – ‘chemical space’ – as several hundreds of thousands conventional compounds;
- Fragments are more efficient in scanning for potential binding pockets on a protein’s surface; and
- Fragments can be readily optimised by growing, linking or merging multiple hits.
Screening using x-ray crystallography provides direct binding information
Due to their small size and low complexity, the binding of fragments to a protein target could be weak, hence a sensitive method for detection that also provides detailed information on the fragment binding mode would be desired – for this we turn to x-ray crystallography.
While x-ray crystallography has been a valuable tool in drug discovery for the past 2-3 decades, mainly to validate binding of hits identified from biophysical screening assays, applying it as a primary screen is less well established, and would require a more high-throughput setup. This is now made possible by the ‘XChem’ facility at beamline I04-1 of Diamond Light Source, which has implemented a number of automation technologies (Figure 1), allowing screening of up to a thousand fragments in as little as a week.
- Fragments are rapidly dispensed to crystals by sound waves (ECHO acoustic liquid handling)
- Crystals are mounted with the help of the Shifter, a robot-assisted microscope stage
- Data collection and initial processing (Xia2) is entirely automated
- The XChemExplorer software suite – automatically calculate electron density maps, generate protein models (DIMPLE), and scan for potential positives (PanDDA) for all datasets in parallel.
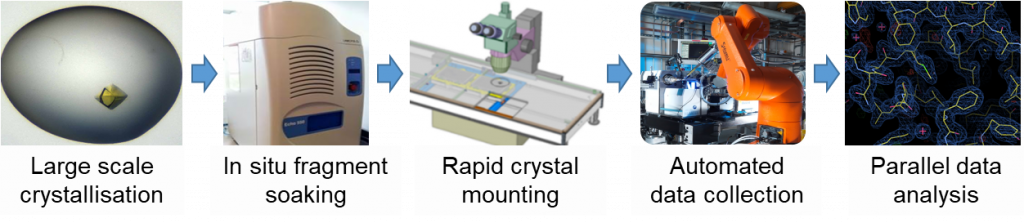
Figure 1: High throughput x-ray crystallography: the ‘XChem’ pipeline.
HAO1 is particularly suited to screening by x-ray crystallography
- Thanks to earlier work here at SGC Oxford in crystallising HAO1, I already have a good start procedure for making crystals for fragment soaking
- Previous crystal structures have provided data on several ligand-dependent conformations of HAO1
My first goal was to find a HAO1 protein construct and crystallisation condition that consistently gave lots (hundreds!) of high quality crystals. From testing a nested series of constructs with varying domain boundaries (figure 2), I found that the HAO11-368 protein readily generates several hundreds of reproducible crystals suitable for soaking.
After collecting high resolution data (1.2 – 1.8 Å) from 409 fragment-soaked crystals, the PanDDA software identified 17 bound fragments, of which five were in biologically relevant pockets of the protein.
You can read an in depth description of how the crystal system was set up and the fragment screen performed, as well as the experimental protocols, in the Zenodo document here.
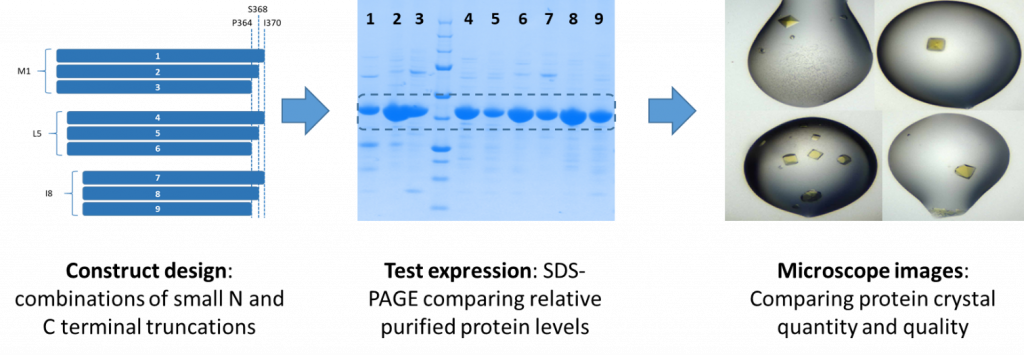
Figure 2: Development of a robust crystal system to support HAO1 fragment screen
The five interesting hits clustered to three sites (figure 3): the active site (fragments 1 and 2), the gating loop at the entrance of active site (fragment 5), and the subunit interface of the HAO1 tetramer (fragments 6 and 7).
In my next post, I will describe more on analysing and triaging the fragment hits for biophysical validation.
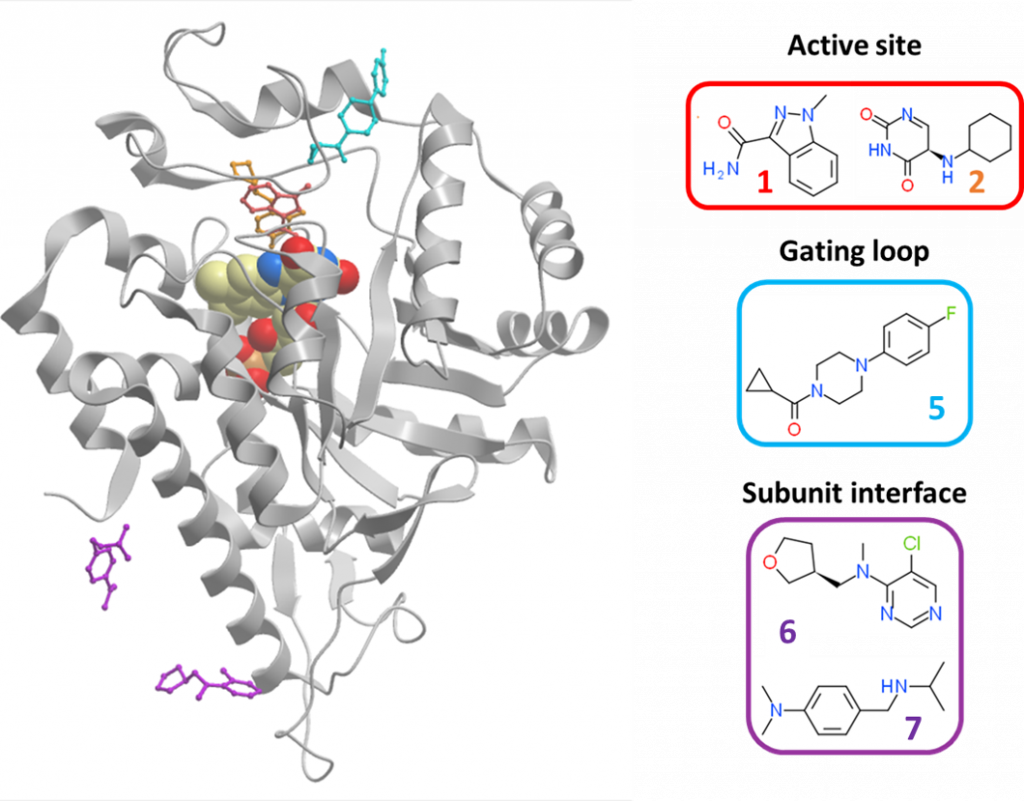
Figure 3: Overview of interesting HAO1 fragment hits. Active site hits are in red; gating loop hit is in blue; and subunit interface hits are in purple.
One Reply to “Identifying fragments that bind to HAO1 by x-ray crystallography”