Synopsis
The four, structurally similar Mur ligases are essential for the survival of gram-negative bacteria and are therefore potential targets for new antibiotics. Crystallographic fragment screening may provide new avenues for inhibitor development.
Background
The rapid emergence of bacterial resistance to almost all clinically available antibiotics is one of the biggest healthcare challenges of the 21st century. The biosynthesis pathway of the peptidoglycan (PG) layer of the bacterial cell wall has proven to be a suitable target for clinically relevant antibiotics. PG biosynthesis starts in the cytoplasm of gram-negative bacteria with MurA and MurB generating the initial precursor UDP-N-acetyl muramic acid. The four structurally similar, ATP-dependent Mur ligases (Mur C,D,E,F) catalyze the stepwise addition of a pentapeptide to the precursor before it gets translocated across the plasma membrane (Kouidmi et al., 2014). All four enzymes are essential for bacterial survival and there was initially great hope that they might serve as targets for new antibiotics. Unfortunately, Mur ligases met the same fate as most other targets for new antibiotics of the high-throughput (HT) screening era: none of the identified compounds ever made it into a clinical program (Silver, 2013). So, the title above really deserves a huge question mark at this point!
Nevertheless, we believe that there is some scope for at least compound development due to the availability of the XChem fragment screening platform. Fragment based drug discovery is a viable alternative to HT screening (Erlanson et al., 2016). The possible number of drug-like compounds with up to 30 heavy atoms is astronomical, whereas fragments contain typically less than 20 non-hydrogen atoms, which reduces the number of possible compounds enormously. Hence, a fragment library with just a thousand compounds samples chemical space a lot more efficiently than a HT library with millions of compounds and because of their smaller size, fragments exhibit reduced molecular complexity: larger compounds can form more interactions, but that means that even just a single, ‘wrongly’ placed atom can ruin an otherwise perfectly fitting compound. Fragments form fewer interactions and therefore have almost always far higher hit rates than HT screens, because they sample the binding properties of a protein more comprehensively. And fragment screens often produce not just a single lead, but provide a diverse set of starting points. The drawback is that fewer interactions means reduced affinity, which in return means binding of a fragment is often more challenging to detect. Unfortunately, high compound concentrations interfere with most assays. Fragment screening by X-ray crystallography does not have this problem; here only the solubility of the compound is limiting. Plus, screening by X-ray crystallography allows immediate visualization of the protein-fragment interaction, making false-positive hits virtually impossible. The reason why it was so far rarely used as a primary screening method is because of its limited throughput.
Mur ligases consist of two domains which undergo significant domain movements upon ATP binding (figure 1), and which has a huge effect on their binding properties (figure 2b,c). Poor cell penetrance of inhibitors may only partly explain why most compounds fail when tested in vivo. Another reason could be that they actually compete with high intracellular concentrations of substrates or cofactors. Most structure-based drug design projects of Mur ligases focused on compounds targeting the ATP binding site. Therefore, targeting secondary or allosteric binding sites could circumvent this problem. Crystallographic fragment screening makes it easy to detect secondary binding sites (figure 2a) (Ludlow et al., 2015). Mur ligases are highly flexible, with the shape of their surface changing considerably during the catalytic cycle (figure 1). Any drugs binding outside the substrate or ATP binding pocket will not compete with endogenous ligands, but will nevertheless interfere with ligase activity.
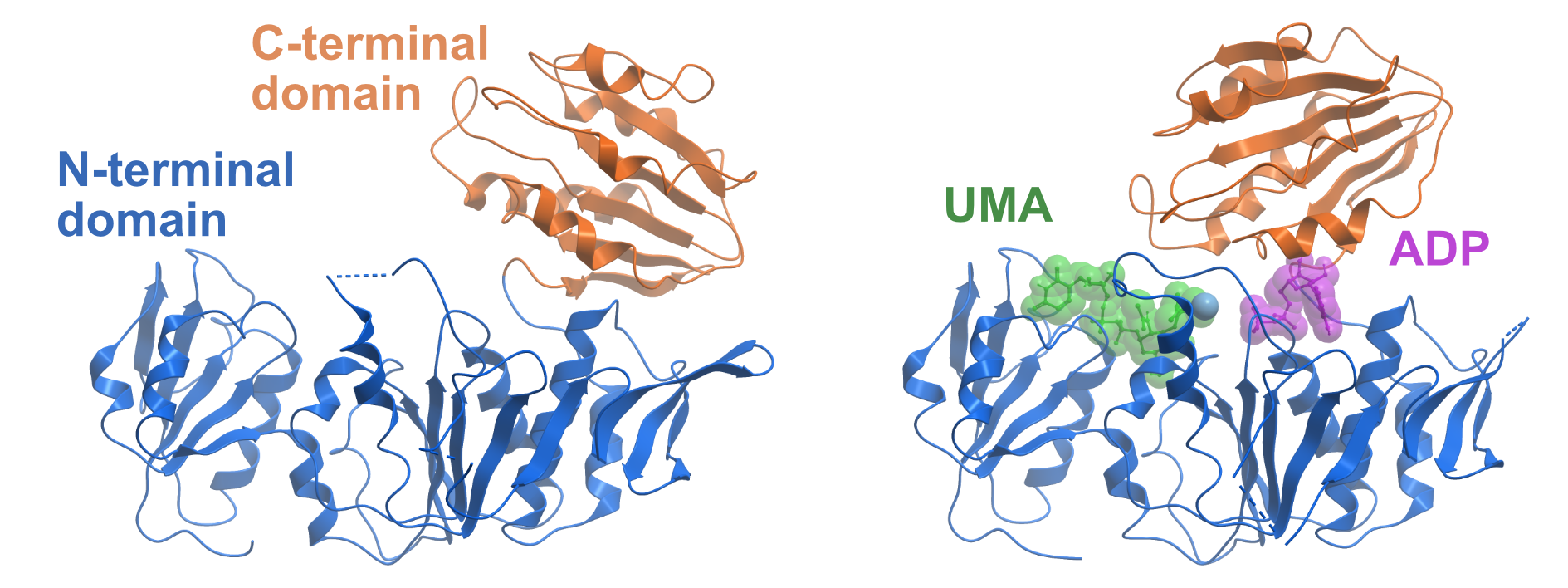
Figure 1. Domain movements of MurD. The left side shows the apo-structure of MurD from Escherichia coli (PDB ID 1E0D). The N-terminal domain is coloured in blue and the C-terminal domain is coloured in orange. The right side displays MurD in the presence of ADP and the substrate Uridine-5′-Diphosphate-N-Acetylmuramoyl-L-Alanine (UMA) (PDB ID 3UAG). The C-terminal domain undergoes a large movement upon ATP binding and thereby closes the ATP binding site
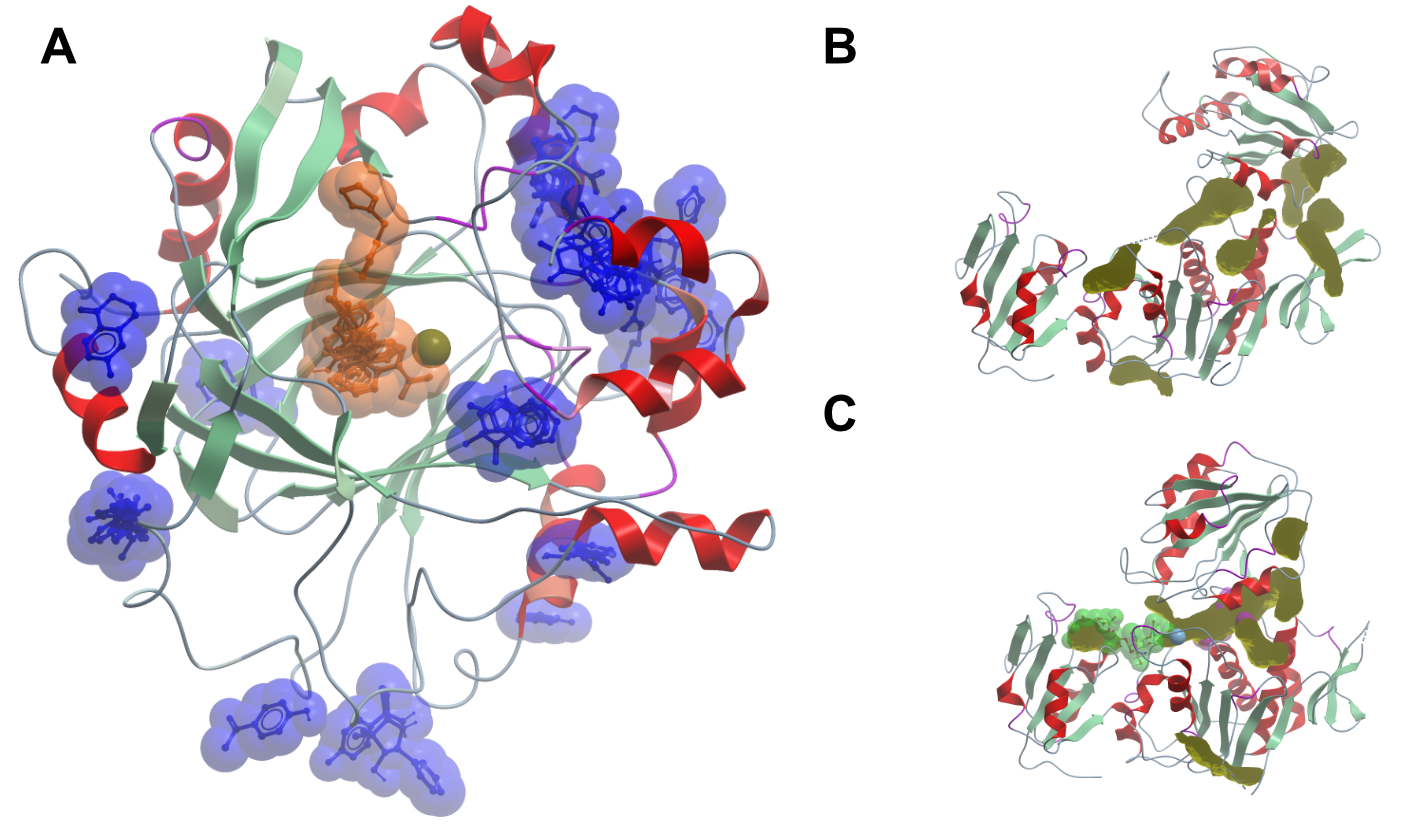
Figure 2. Experimentally determined and predicted fragment binding sites. The left side shows the results of a crystallographic fragment screen on human KDM4D (A). The picture shows an overlay of all compounds binding to the protein; compounds binding at crystal contacts were excluded. Compounds binding to the active binding site are coloured in orange, all other binding compounds are coloured in blue. The right side of the panel shows MurD in the open (B) and closed (C) conformation. The binding pockets in yellow were calculated with the icmPocketFinder (Molsoft LLC) method. The reliability of the algorithm is not clear, but it illustrates the vastly different surface properties of the two states of the same protein.
Workplan
Clone, express and purify MurC, MurD, MurE and MurF from Escherichia coli. Then perform crystallographic fragment screening at the XChem facility at the Diamond Light Source.
Current status
We have already some results, but we are unfortunately a bit behind in documenting the progress of the project on Open Lab Notebooks. Additionally, we have in the meantime teamed up with Chris Swain (Cambridge MedChem Consulting), Matthew Todd (UCL) and Chris Dowson (Warwick University) in order to help the project progress more quickly. Chris and Matthew have created a platform on Github to invite anyone with an interest in open source drug discovery to throw in ideas: https://github.com/opensourceantibiotics. Also, structures can already be viewed and downloaded via FRAGALYSIS. Nevertheless, we will provide more detailed protocols about how the proteins were prepared etc. shortly.
Literature
Kouidmi, I., Levesque, R.C., and Paradis-Bleau, C. (2014). The biology of Mur ligases as an antibacterial target. Molecular Microbiology 94, 242–253.
Ludlow, R.F., Verdonk, M.L., Saini, H.K., Tickle, I.J., and Jhoti, H. (2015). Detection of secondary binding sites in proteins using fragment screening. PNAS 112, 15910–15915.
Munshi, T., Gupta, A., Evangelopoulos, D., Guzman, J.D., et al. (2013). Characterisation of ATP-Dependent Mur Ligases Involved in the Biogenesis of Cell Wall Peptidoglycan in Mycobacterium tuberculosis. PLOS ONE 8, e60143.
Ruane, K.M., Lloyd, A.J., Fülöp, V., Dowson, C.G., Barreteau, H., Boniface, A., et al. (2013). Specificity Determinants for Lysine Incorporation in Staphylococcus aureus Peptidoglycan as Revealed by the Structure of a MurE Enzyme Ternary Complex. J. Biol. Chem. 288, 33439–33448.
Silver, L.L. (2013). Viable screening targets related to the bacterial cell wall. Annals of the New York Academy of Sciences 1277, 29–53.