In my previous post https://openlabnotebooks.org/protein-protein-docking-for-protac-discovery/ I showed that HADDOCK was the best protein-protein docking tool among those I tested to predict how E3 ligases interact with their protein substrates. Here, I ask whether docking virtual libraries of PROTAC candidates to these E3 ligase – substrate protein interfaces can be used to predict which PROTACs are active.
The virtual screening method that I am using is as follows (Figure 1):
- Use HADDOCK to dock the E3 ligase to the protein target and keep the ~40 best solutions (typically grouped in 5 to 9 clusters of structurally related binding poses)
- For each PROTAC candidate, tether the two chemical handles to their corresponding ligands in the protein-protein complexes generated by HADDOCK, and minimize the energy of the PROTAC while ignoring the proteins. This produces a selection of conformationally acceptable PROTACs: SELECTION 1
- Start from the conformationally acceptable PROTACs (SELECTION 1) and further minimize their energy, this time accounting for the surrounding proteins (PROTACs and side-chains within 5Å are kept flexible)
Step 2 and 3 are conducted 3 times independently, and all results are merged, so, for 10 PROTAC candidates, we will have 10 molecules x 40 prot-prot-interfaces x 3 repeats = 1200 binding poses.
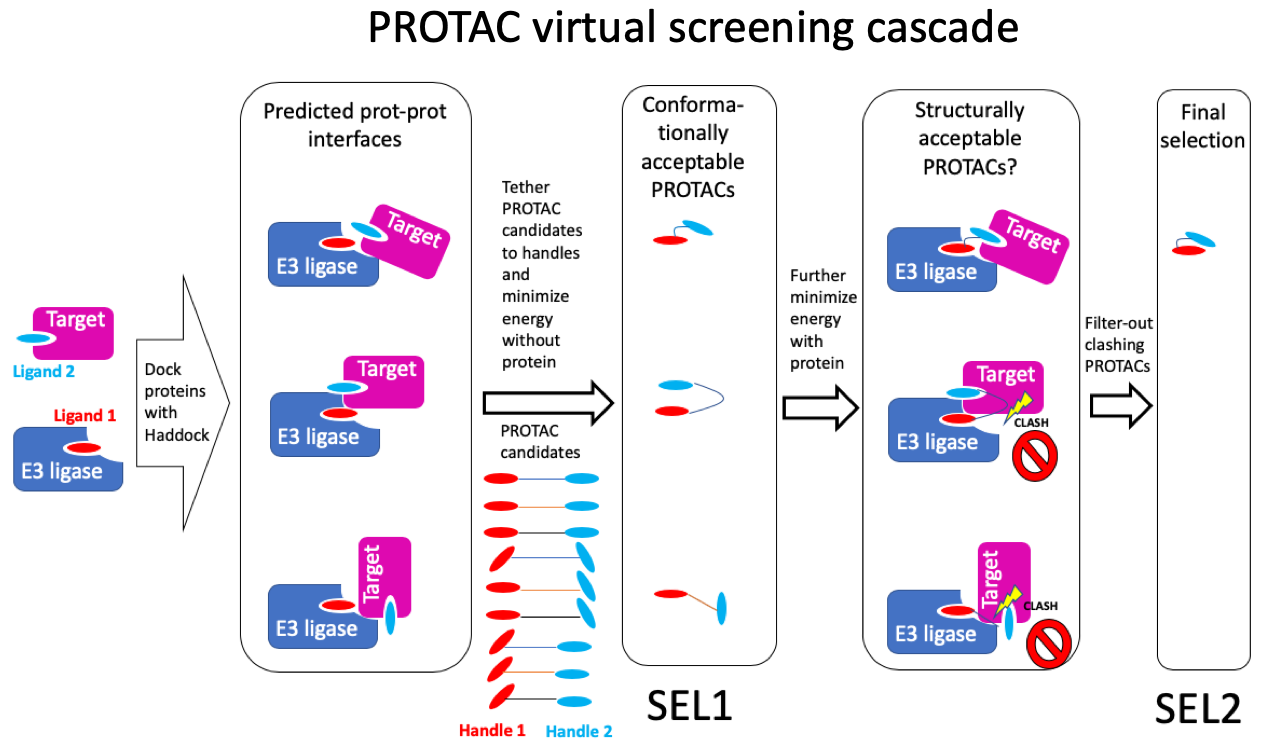
Figure 1: Overview of the virtual PROTAC screen pipeline.
This screening pipeline is implemented in ICM (Molsoft, San Diego), and the corresponding script is attached to this Zenodo post.
I tested this protocol against 5 test cases taken from the literature, and the details can be found on Zenodo. Here is a summary table:
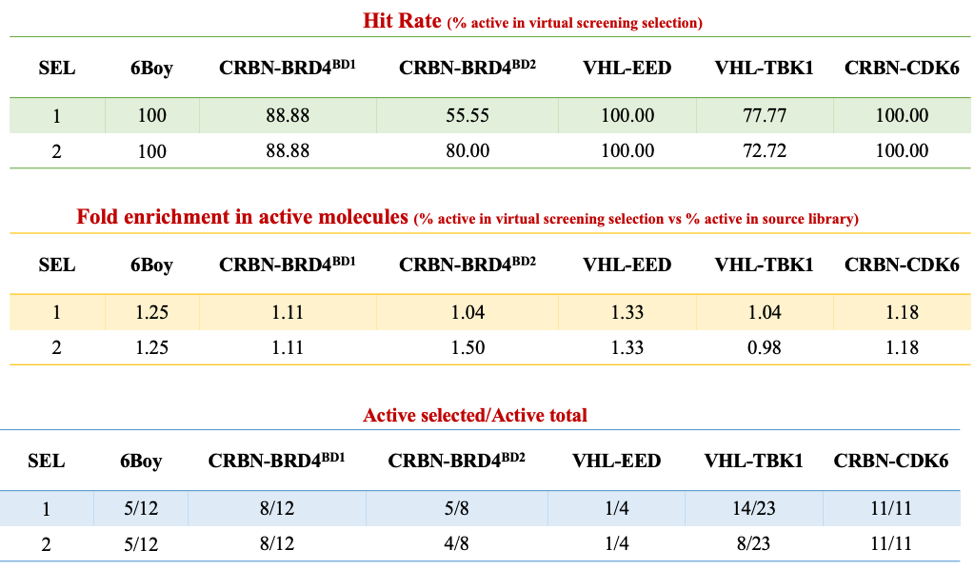
Two general observations can be made. First: in all test cases, the starting library of compounds for which we have experimental data is highly enriched in experimentally active molecules. Picking compounds at random, one has good chances of selecting active PROTACs. Two: in all 5 test cases, our top 3 scoring PROTACs out of the virtual screening pipeline are systematically active (see details in Zenodo). In four out of 5 test cases, the library selected by virtual screening is enriched in active PROTACs, compared with the starting library, which is better than random. At this point, it is too early to know whether this is pure luck, or whether this reflects a valid virtual screening strategy.
Considering how unreliable virtual screening of conventional small molecules to a crystal structure is (one can expect 95 inactives out of 100 compounds selected virtually when the stars are aligned), it seems almost ridiculous to dock PROTACs (large and floppy molecules) to predicted protein-protein interfaces. And yet, it is not unreasonable to expect that the multiple protein-protein docking poses generated by HADDOCK (or other protein-protein docking tools), while being all suboptimal (after all, these complexes do not form naturally), are structurally sufficiently sound to be stabilized by a PROTAC. Indeed, Nowak et al. clearly showed that different PROTACs could induce different binding poses between CRBN and BRD4BD1 (Nowak et al. Nat. Chem. Biol. 14:706-714). If we accept this hypothesis, the PROTAC docking step should actually be more efficient than a regular virtual screening campaign, as the orientation of the two functional ends of the PROTACs are imposed by the protein complex, which considerably reduces the conformational space available to the PROTAC, and the chances to predict a wrong PROTAC binding pose. There is still room for getting things wrong, but not as much as one would expect considering the complexity of the system.
Finally, an important factor I ignored so far is that some PROTAC candidates may induce the formation of a ternary complex in a biophysical assay, but still be inactive because they do not cross cell membranes. I will probably need to incorporate physico-chemical filters to account for this, following-up on work by Maple et al. ( MedChemComm., 2019, DOI: 10.1039/c9md00272c) and Edmondson et al.(Bioorg. Med. Chem. Lett., 2019, 13:1555-1564) Another factor even more complex is that some compounds may induce the formation of a ternary structure that is not compatible with efficient ubiquitylation of the target.
Two things are sure: we need more experimentally inactive PROTACs, and there is room for improvement.