Our initial fragment screen identified four different fragments that bound to what we call site F (Figure 1) These can be found in the Protein Data Base (pdb) with these codes: 5QSA, 5QSC, 5QSI, and 5QSK. In this blog I will talk about some observations from these structures that suggest promise for this pocket, show two structures (lactam and indoline acid) that came from a virtual screening exercise using this pocket, and finally present new data from a series of lactams we have synthesized and purchased to begin to explore the pocket more carefully. 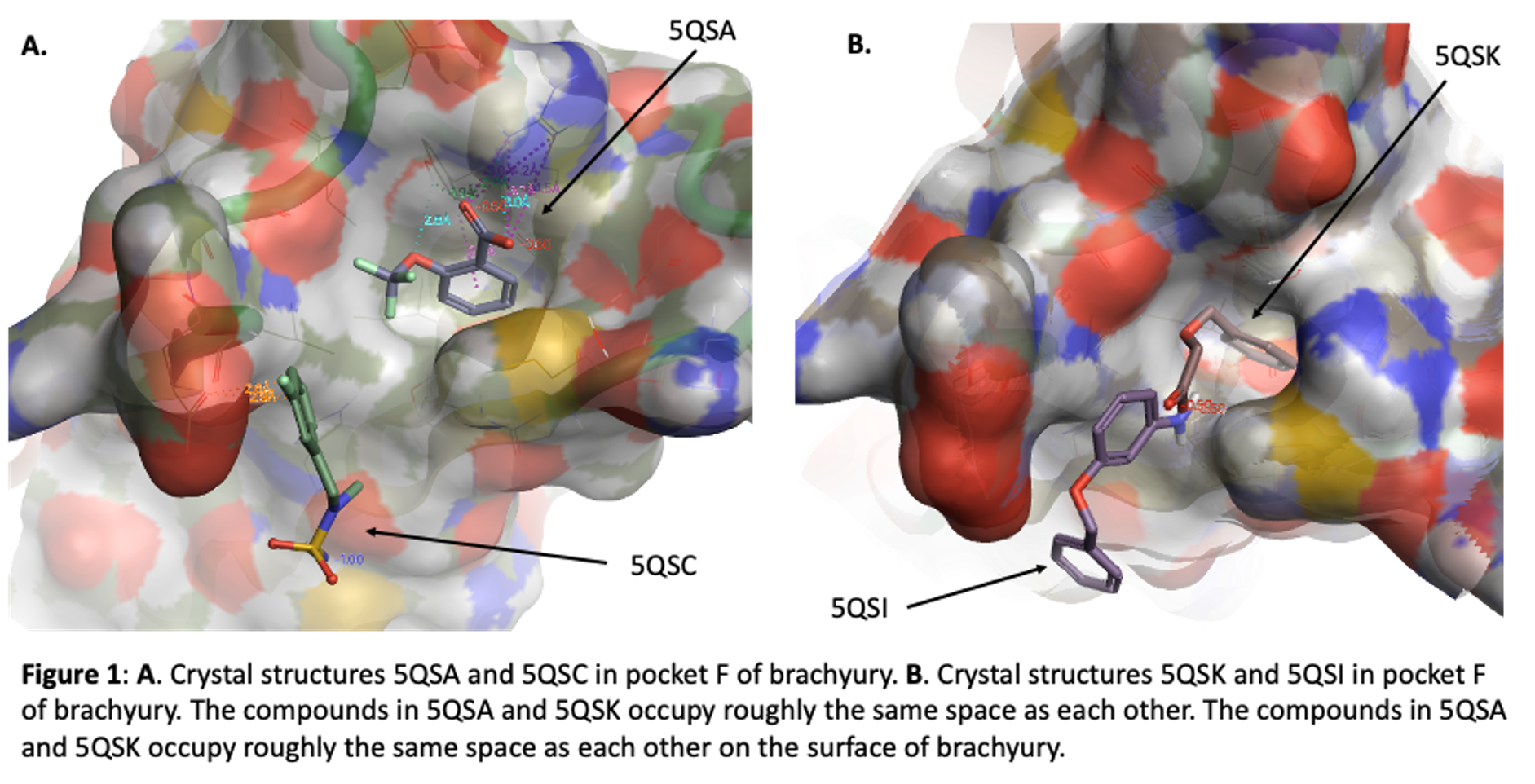
The ortho-OCF3 benzoic acid (compound FM001580, pdb 5QSA) is buried in pocket F. The phenyl ring makes an edge to face interaction with TYR88 of TBXT and hydrophobic interactions with the side chain of Met181. The carboxylic acid interacts with Arg174 and also with the phenol of TYR88. The ether oxygen of the ligand makes a hydrogen bond with the phenol of TYR88. It appears that one could grow from the OCF3 vector and build new interactions along the protein surface. The aromatic ring of another carboxylic acid (FM002150, pdb 5QSK) binds in the same hydrophobic pocket but the carboxylic acids of these two ligands do not overlap. Instead, the acid of FM002150 extends towards solvent. This is along the same vector as the OCF3 of FM001580, supporting this as a promising position for new analogues. Interestingly, ARG174 moves in the second structure, modulating the shape of the pocket.
Benzyl ether aniline FM001452 (pdb 5QSI) and sulfamide F9000560 (pdb 5QSC) also bind in the F site but they are offset from the position of the two carboxylic acids. These two compounds occupy similar space to each other in the brachyury protein. They interact with Glu41 and bound water molecules.
So, we have 2 pairs of ligands in site F, slightly offset from each other. In the first pair a phenyl ring makes key interactions with TYR88 and MET181, and ARG174 can be accessed. The other pair is not quite so buried but nonetheless appears to be a potential hot spot for binding, with interactions with Glu141. Together these data suggested that larger compounds could bind in site F and take advantage of features of both of these off-set ligand locations, perhaps leading to enhanced potency. We embarked on two strategies to explore site F. The first involves purchase and synthesis of analogues that explore these 4 cores. These incorporate features that take advantage of the observed key binding elements and grow towards the new opportunities. These compounds will be subject of a future post. Here I will talk about hits from a virtual screening exercise.
Louisa Temme (former postdoc in SGC-UNC) collaborated with Tigran Abramyan (former postdoc in the group of Alex Tropsha, a computational chemist in the UNC Eshelman School of Pharmacy) to perform a virtual screen. We hypothesized that the size, shape, and composition of pocket F is conducive to potent ligand binding if we can take advantage of the pocket shapes and interactions. In brief, the virtual screening involved docking of large numbers of compounds into this site. Tigran docked the Enamine 500k diversity set supplemented with commercially available compounds we identified based on substructure and similarity searching around the four crystallized ligands identified in our initial crystallography screen. High scoring hits were clustered based on their structures and inspected by the team. We looked for compounds that were buried in the site like the crystallized starting points, formed (in the docking visualization) similar interactions with TBXT as the initial crystallography hits. Purchased compounds were soaked into the TBXT crystals and two new structures were solved, and the compounds indeed bound in site F.
The two series are an indoline acid and a lactam. They are depicted in figure 2 along with the images of hem bound to TBXT in pocket F. Both take advantage of the lipophilic pocket that was identified in 5QSA and 5QSK although they fill the space in slightly different ways.
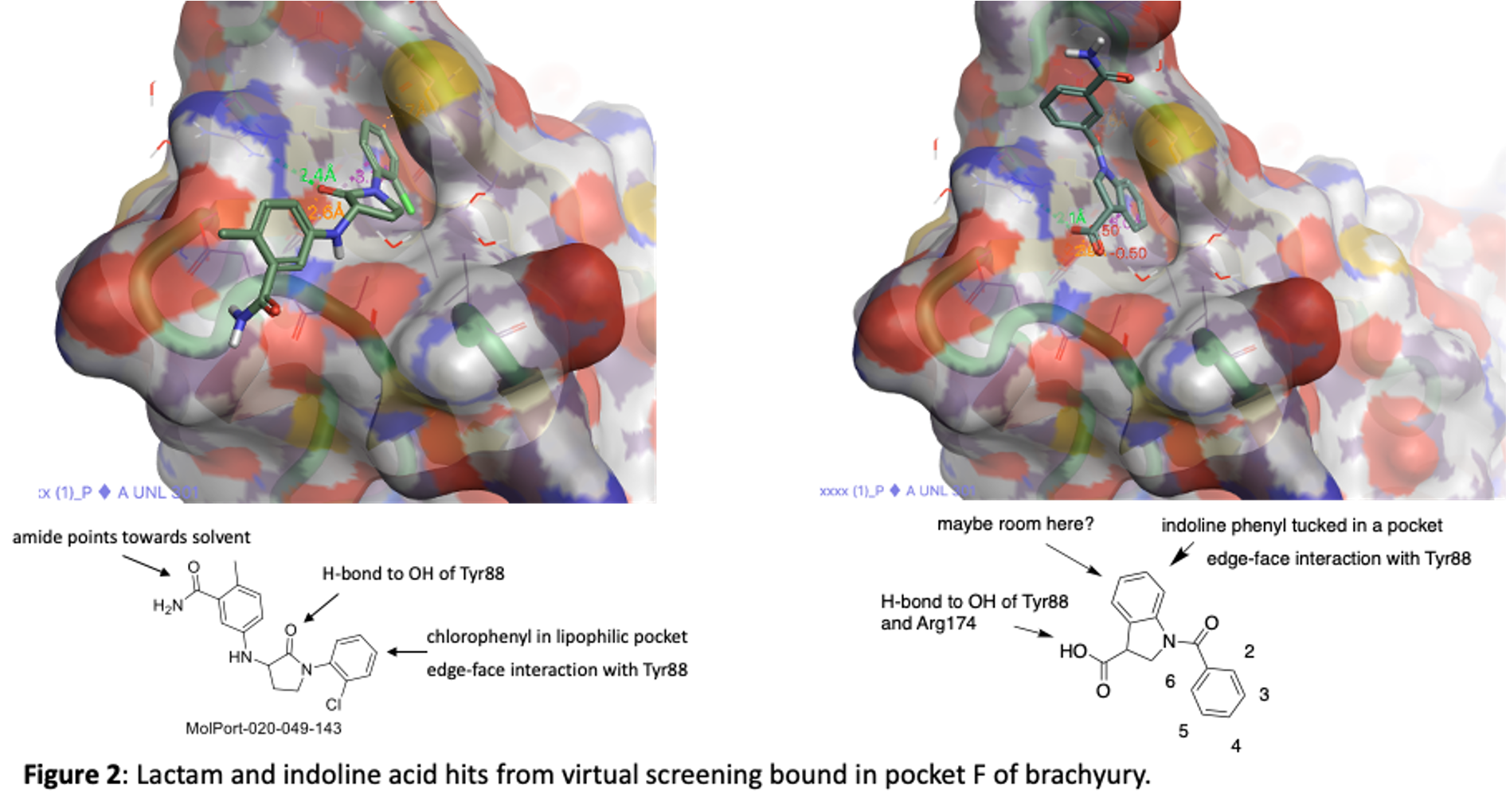
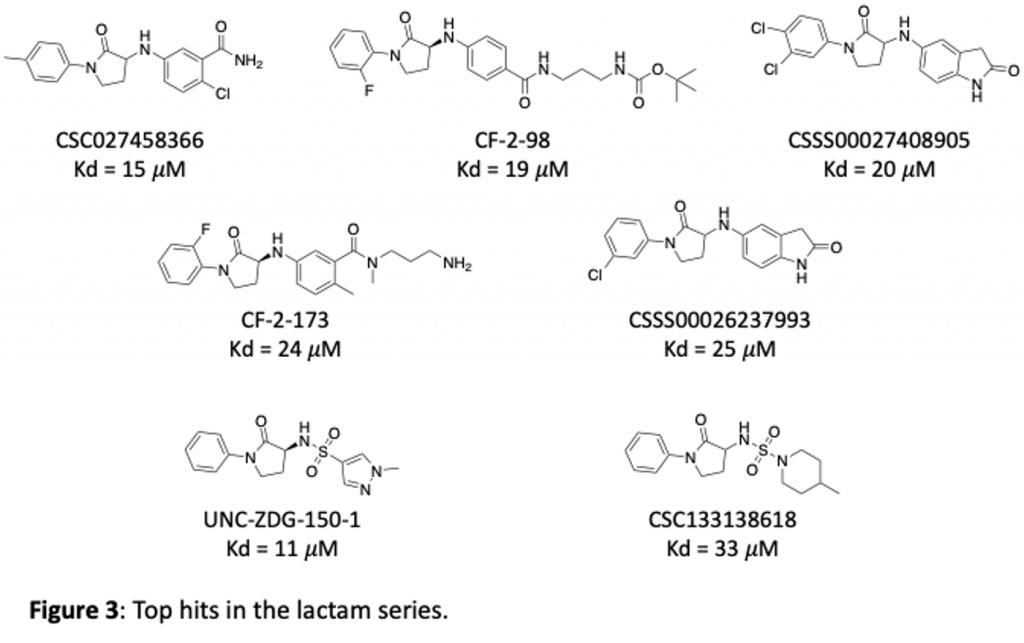
We have purchased and made a number of these now and tested them in our SPR binding assay to measure their affinity for brachyury. Some of the most potent lactams are shown in figure 3. Thanks to Zach and Anwar and the chemistry team at Piramal for work on this project!
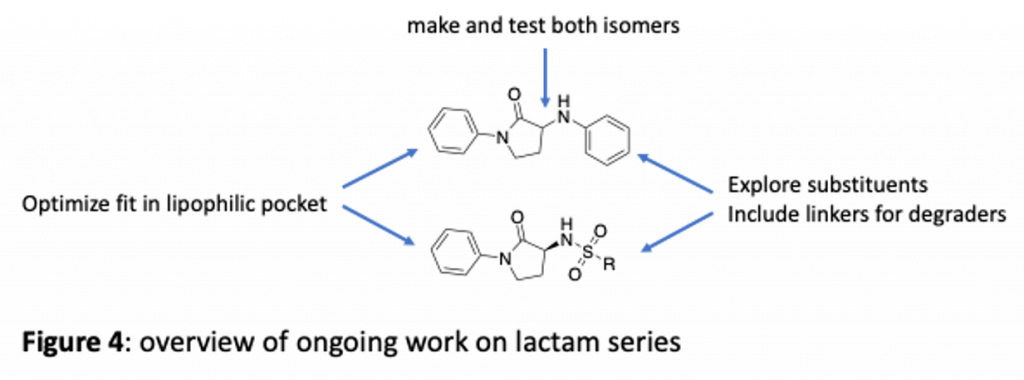
The lactams are a high priority series and we are making a variety of changes (overview in figure 4). For example, we are exploring the phenyl that projects into the lipophilic pocket to see if the fit can be optimized. We are also looking at further changes to the pendant aniline moiety to try to make new interactions. This is also a potential site for linkers to make reagents that may be able to degrade brachyury. You can see we have also explored sufamides, exemplified by ZDG-150-1. Promising activity here means we will explore more sulfamides as well.
As always, feel free to ask questions. We will be posting on other promising series soon.
Thank you for the update. I share each of theses posts with the Chordoma Survivor community on FB, though we know you are a long way from a clinical application, your updates buoy our spirits and give us hope for better treatments in the future. Thank you again from all those also touched by Chordoma.
Dear John,
You are very welcome. Thank you for sharing these posts with your community and also for letting me know that they are helpful to you! I am grateful for the opportunity to work on finding a treatment for chordoma and am inspired by people like you in the patient and caregiver community!
Take care,
David