The year is coming to an end and I want to start my blog by giving you an overview of a project that kept me busy for the better part of it. Our existing Protein Data Bank (PDB) batch deposition mechanism underwent a major overhaul recently and we are finally in a position where we can relatively quickly deposit all the structures from an XChem fragment screening campaign.
I assume not everyone will be familiar with what this is about, so here is some background of the problems we are trying to solve: once the XChem facility started churning out more and more protein-fragment complex structures, it became obvious that we needed new tools to support PDB deposition. Doing it one-by-one through the wwPDB website would be too time-consuming. Additionally, the multi-dataset nature of the experiment and the way we analyse the data made it necessary to think about new ways to help the end-user understand the basis of the modelling decisions taken by the depositor.
Summary
The post is rather long, because it contains quite a few details which might be of interest if you are confronted with having to deposit many similar structures or if you want to publish findings based on a PanDDA analysis. Here’s a quick summary if you are in a rush:
- Each deposited structure contains direct evidence for the modelled ligand, i.e. the PanDDA event map
- All apo structures that were collected during an XChem experiment are deposited as a single PDB entry so that the data can be used to reproduce the PanDDA analysis
- File preparation for batch deposition of ligand bound and apo structures is facilitated by XChemExplorer
PDB group depositions
The first problem, i.e. deposition of many structures, was solved quickly, because, fortunately for us, the PDB has already developed a new, but not very well known, portal to support parallel structure depositions, so-called group depositions. It’s pretty nifty, you can bundle all your structure and structure factor mmcif files in a single tar archive, then you upload the file and press submit. That’s it! Well, almost. One still needs to provide the usual experimental data like protein sequence, crystallization condition, authors etc. in the header of each mmcif file, but I’ve implemented a solution in the XChemExplorer (XCE) software so that one only needs to do it once and the information then gets propagated to all the structures in a group (Figure 1). Once the user has provided this information in XCE, they need to flag which structures they want to deposit and the program will generate the respective files, ready for upload. That was the – conceptually – easy bit. Next, we had to think about ways to provide evidence for the modelled ligands.
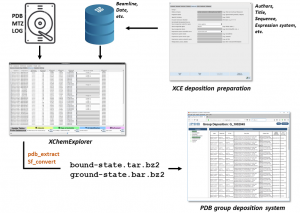
Figure 1. XChemExplorer (XCE) enables preparation of files for mass structure deposition through the PDB group deposition portal. Users only need to provide a minimal amount of information which is then propagated to all files in a group. The remaining information is captured from the XCE database and the project directory. XCE uses PDB_EXTRACT and SF_CONVERT to generate structure and structure factor mmcif files, respectively. XCE can generate and bundle mmcif files of ligand-bound models, as well as of the ground-state model and the corresponding apo structure factor mmcif files.
Evidence for modelled ligands
We routinely use the Pan-Dataset Density Analysis (PanDDA) method to identify bound fragments and because the algorithm is so sensitive, we are often able to detect weakly bound compounds which would go unnoticed in 2mFo-DFc maps. However, the concept of PanDDA event maps is still just emerging in the community and so the question is how can we provide the rest of the world with evidence that these ligands really are bound? The last few years have seen a few efforts to improve the quality of protein-ligand structures in the PDB and there have been several publications which highlight dubiously modelled ligands. So, what to do in order to avoid that XChem will soon find itself in the pillory? Together with Stephen Burley’s team at the RCSB, we enabled inclusion of PanDDA event maps in the deposited structure factor mmcif file (Figure 2). This allows everyone – with a bit of crystallographic knowledge – to download the mmcif file and recreate the maps that were used to model the bound ligand and eventual changes in the protein structure. Ideally, the event maps would be displayed on the different PDB websites, but in the meantime, we have created our own interactive HTML summaries which users can use to share their results with the world. On a personal note, I would like to add that there certainly are ‘fantasy’ models in the PDB, but this is probably unavoidable given the sheer number of structures in the archive and that overall, protein crystallographers are most likely not too dissimilar to the rest of humanity when it comes to the distribution of certain traits. Still, I am sure that most scientists are aware of the limitations of some of their models, but they have no possibility to communicate their concerns. However, this is another topic which I will elaborate on in one of my next blog posts.
Reproduction of results
Finally, we also thought about ways to enable anyone to reproduce the event maps. Ultimately, PanDDA event maps cannot be reproduced unless you have all the apo datasets available to calculate the respective statistical maps. Making all the data available, also ensures that PanDDA users can be confident when publishing their results. Technically, this should be trivial in the year 2018 given that the file sizes involved are negligible by today’s standards. The more important (and contentious) question is ‘where should the data live?’. The PDB recommended to deposit them in their archive so that they will be properly curated and remain available in perpetuity. Hence, our initial strategy in 2017 was to bundle all the apo structures in another tar archive and deposit them in a separate group deposition session. But that meant that every apo structure was treated as a unique structure and therefore got its own PDB ID. Needless to say that this created confusion in some parts of the community who were wondering what we are up to. This did clearly not work as expected, not least because even if someone would like the run PanDDA again on the data, they would need a lot of patience if they want to download all the datasets from the PDB. We contemplated to use the ZENODO data repository as an alternative (and we still recommend to use this platform to upload the complete PanDDA analysis anyway), but finally we came up with a, which at least I believe, much more elegant solution. Instead of depositing every apo PDB file (which are almost identical after initial refinement), we deposit only the ground-state model (and the corresponding mean map) and then include the structure factors of every dataset that was collected during the XChem experiment in the final structure factor mmcif file (Figure 2). The file will often be several Gb large, but users will now be able to download all the data that they need to reproduce a PanDDA analysis in one go.
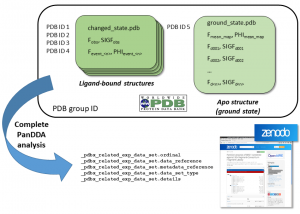
Figure 2. File structure and contents of the deposited mmcif files. Each ligand-bound structure (changed_state.pdb) does consist of an mmcif file containing its coordinates and experimental data and a structure factor mmcif file which contains the measured data as well as an additional data block for each PanDDA event map. The ground-state model will be included in a separate mmcif file. The corresponding structure factor mmcif file does contain one block for the mean map and then one block each for every datasets which was part of the XChem experiment. In case someone wants to reproduce the PanDDA analysis, they can download the ground-state deposition, use the model to run initial refinement on every dataset and then subject it to PanDDA analysis. Additionally, there is a possibility to upload the complete PanDDA analysis into the ZENODO data repository and point to it in the PDB deposition.
We have recently made our first apo deposition (G_1002058) and are still sorting out a few technicalities. Once this is done, I’m planning to make a few changes to the XCE gui, so that users get a simple step-by-step description what they need to do. Also, I am currently working on a publication which we will hopefully submit early next year and which will provide more details.