Hello again! A few posts ago (here), I presented one fragment bound at the interface between two subunits of the HAO1 tetramer (figure 1 will refresh your memory). Our idea to optimize this fragment into an inhibitor would be to make it bigger so that it disrupts the HAO1 tetramer. I would expect this to reduce HAO1 activity either directly (if HAO1 is more active as a tetramer and/or the compounds change the conformation of the tetramer), or indirectly, as disrupting the tetramer could reduce the stability of HAO1 protein.
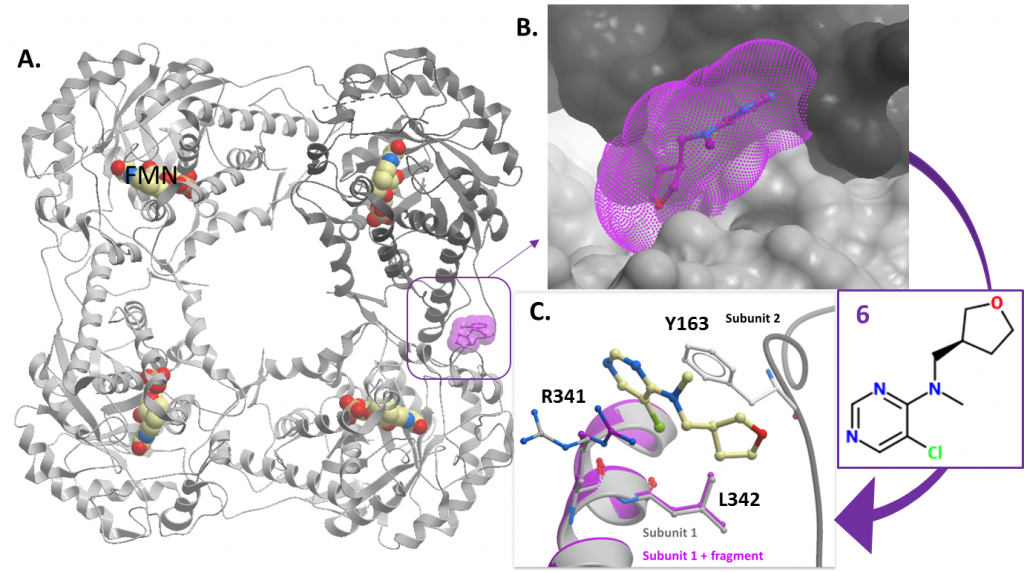
Figure 1: Fragment 6 binds at the interface between two subunits of the HAO1 tetramer. A. Location of fragment binding in relation to the HAO1 tetramer. B. Surface model showing fragment binds in pocket between two subunits. C. Ribbon and stick model showing fragment 6 interacts with both subunits, displacing R341 (subunit 1) and pi stacking with Y163 (subunit 2).
With help from Tamas Szommer in Paul Brennan’s group at SGC, I synthesized a small set of fifteen bulky compounds based on the fragment scaffold. I wanted to preserve the aromatic ring (called a pyrimidine because of its two nitrogens), which engages in pi stacking with one subunit of HAO1, and the chlorine substituent, which displaces an arginine on the other subunit, to try to ensure that the compounds still bind to the same place, while replacing the five-membered, oxygen-containing ring (called a tetrahydrofuran) with larger substituents.
To synthesize this set of compounds, I employed a common chemical reaction, called nucleophilic aromatic substitution, to replace one chlorine in a dichloro-pyrimidine with the amine (-NH) group of various building blocks. I chose 15 compounds that were diverse in structure and greater than 150 Daltons in size (a tetrahydrofuran is 72 Daltons) to make my building blocks. In this reaction (shown in figure 2), a nucleophilic group (in this case the amine of the building block, R1-NH-R2) is added to an aromatic ring, delocalising the electrons and forming an intermediate known as the Meisenheimer complex. Then, the halogen atom (in this case the second chlorine) is eliminated from the ring, allowing it to return to its preferred, lower energy, aromatic conformation.
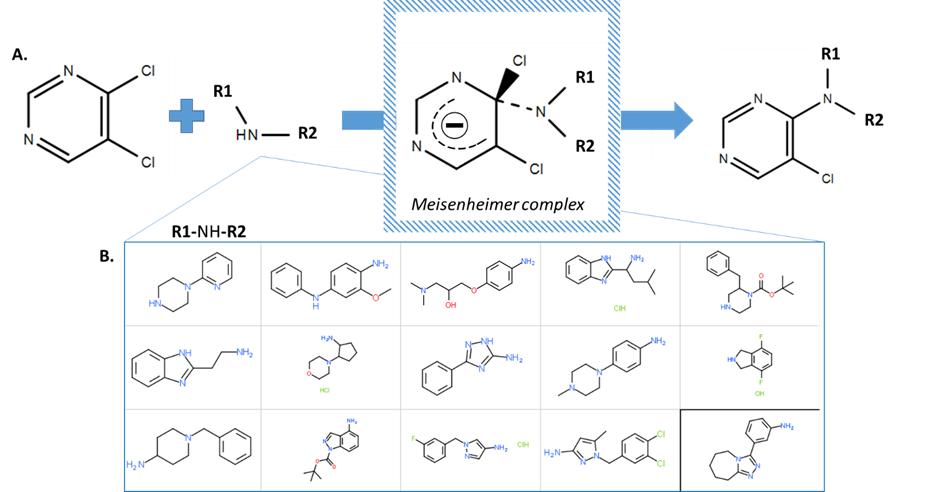
Figure 2: SNAr reaction mechanism and amines used to synthesise follow up compounds. A. SNAr reaction scheme. Inset: Meisenheimer complex intermediate. B. 15 building blocks selected based on size, diversity and inclusion of a free amine (NH or NH2).
I first tested my new compounds for inhibition of HAO1 using my fluorescence-based activity assay (as described in my last post). Of the 15 compounds, only three inhibited more than 50% of HAO1 activity at 1 mM, allowing IC50 determination. The best compound, SM001-10, has an IC50 of 86 uM. Some compounds showed no inhibition at all at 1 mM and this is also useful in determining how (not) to expand the ‘good’ compounds. More details about the synthesis and assaying of these compounds can be found on my Zenodo post here.
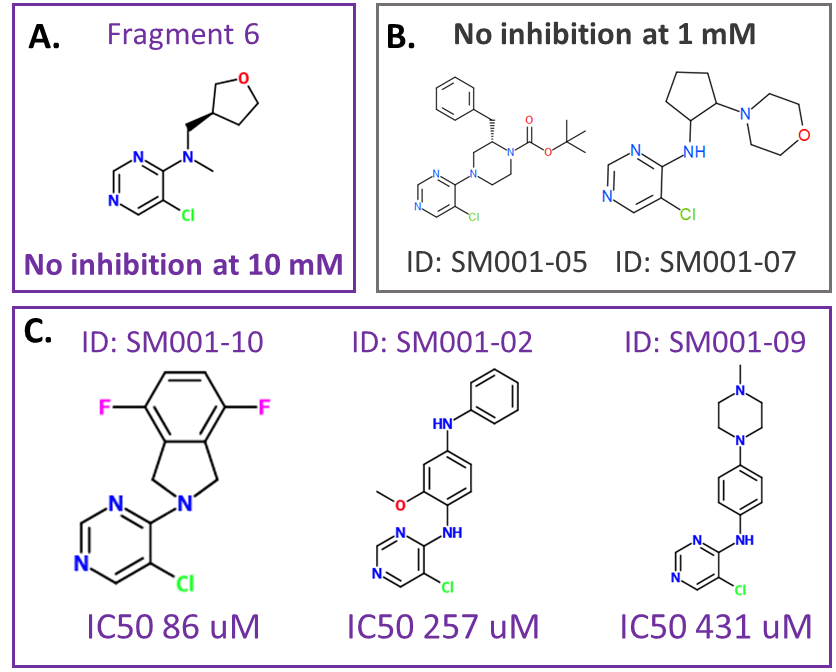
Figure 3: Effect of fragment 6 and related compounds on HAO1 activity. A. Original hit, fragment 6, structure and activity. B. Extensions that did not result in inhibition of HAO1. C. Structure and IC50 results for the three compounds showing significant inhibition of HAO1 at less than 1 mM concentration.
We need two more pieces of information to guide the development of these compound towards inhibitors of HAO1: (1) x-ray crystal structures to tell us how exactly the compounds bind and what residues nearby we could expand the compounds towards to improve their activity; and (2) an assay that tells us more about how these compounds are inhibiting HAO1 activity i.e. do they disrupt the HAO1 tetramer? Or change the shape of the tetramer so that it is less effective at turning over the substrate?
I will tell you more about these in the New Year, until then – Happy holidays everyone!
Very exciting step from an inactive Xray fragment hit to a microM inhibitor. Congrats!