Background
What is the link between a rare genetic disease that causes soft tissues to turn to bone, and a lethal childhood brain cancer? At first glance, with such different clinical phenotypes, it seems unlikely there could be any link, but it has been shown through whole genome sequencing of diffuse intrinsic pontine glioma (DIPG) patients in the past few years that in actual fact, the same mutated protein that causes fibrodysplasia ossificans progressiva (FOP), is present in 27 % of DIPG cases.
FOP is caused in 97 % of cases by a single mutation in the gene encoding for Activin A receptor Type I (ACVR1, also known as ALK2). The disease is usually diagnosed in childhood, where minor trauma to soft tissues causes inflammation followed by heterotopic ossification (HO) of those soft tissues, and gradually over time the patient grows a “second skeleton” from hardening of soft tissues into bone. There is no cure for FOP, and current therapies are symptomatic.
DIPG is the leading cause of death from paediatric brain tumours. The tumours occur in the ventral pons of the brain (the brainstem), making removal by surgery impossible. Radiation therapy can marginally increase lifespan, but for most children with DIPG, survival time is approximately 9 months from diagnosis. No chemotherapeutic agents have been found to slow or halt disease progression. In addition to mutations in ACVR1 in about a quarter of cases, DIPG patients have a K27M mutation in histone protein H3, resulting in changes in gene expression that ultimately lead to tumour formation.
ACVR1 is a membrane-bound receptor that sits in the plasma membrane of the cell and forms a heterotetrameric complex with Type II receptors when bone morphogenetic proteins (BMPs) bind. Both Type I and II receptors have serine/threonine kinase domains intracellularly, and when BMPs bind, the Type II receptors phosphorylate the Type I receptors, thus activating them (see Fig 1).
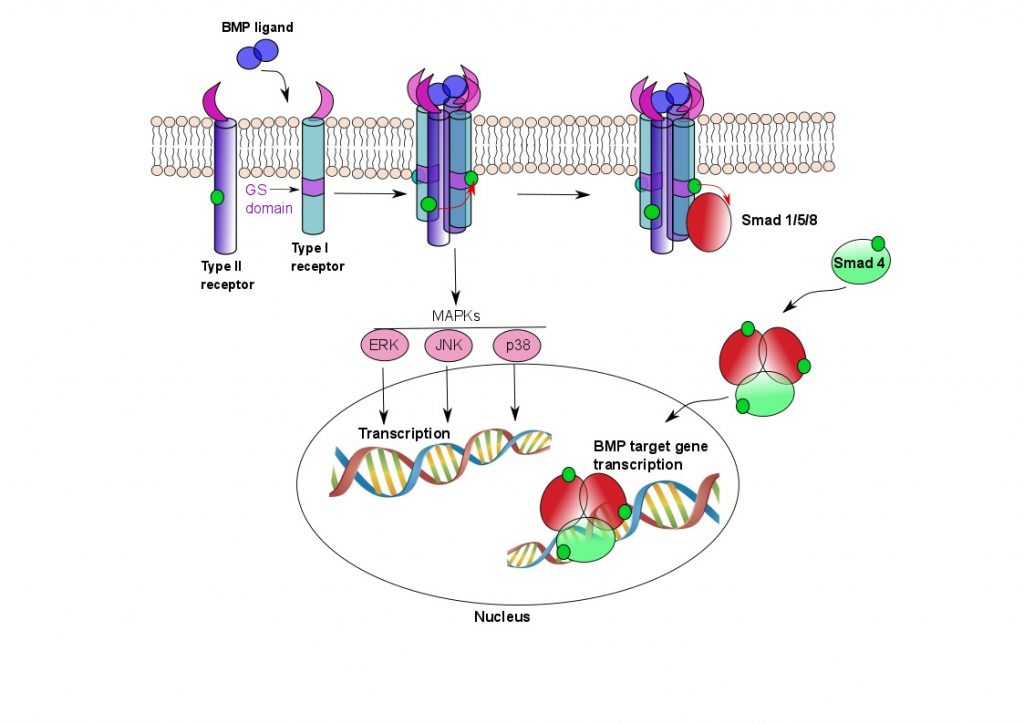
Figure 1. ACVR1 signalling pathways.
Two Type I and two Type II receptors tetramerise upon binding BMP ligand. The Type II receptors phosphorylate the Type I receptors in their GS domain, whereupon the Type I receptors are able to phosphorylate Smad proteins. The Smad proteins oligomerise and translocate to the nucleus, where they activate BMP target gene transcription. BMP signalling results in ectopic bone formation, but is also responsible for a number of other functions, such as maintenance of adult tissue homeostasis. The MAPK pathway is also involved in osteoclast differentiation.
The FOP R206H mutation, also present in DIPG, is in the so-called GS (Glycine-Serine-rich) loop of the kinase domain of ACVR1, where the amino acid Arginine is replaced by Histidine, an amino acid with very different properties (Fig 2). This causes the protein to bind less tightly to its inhibitor protein FKBP12, and to exhibit altered binding activity of activin, which acts as a competitor to BMPs, and, in FOP, ultimately results in HO. Other mutations in FOP, in DIPG and in both diseases, are shown in Figure 2, but the mutations in DIPG occur only in somatic cells, not germline as in FOP. It isn’t known how these mutations interact with the histone mutations to cause oncogenesis, but ACVR1 mutations on their own very rarely cause cancer.
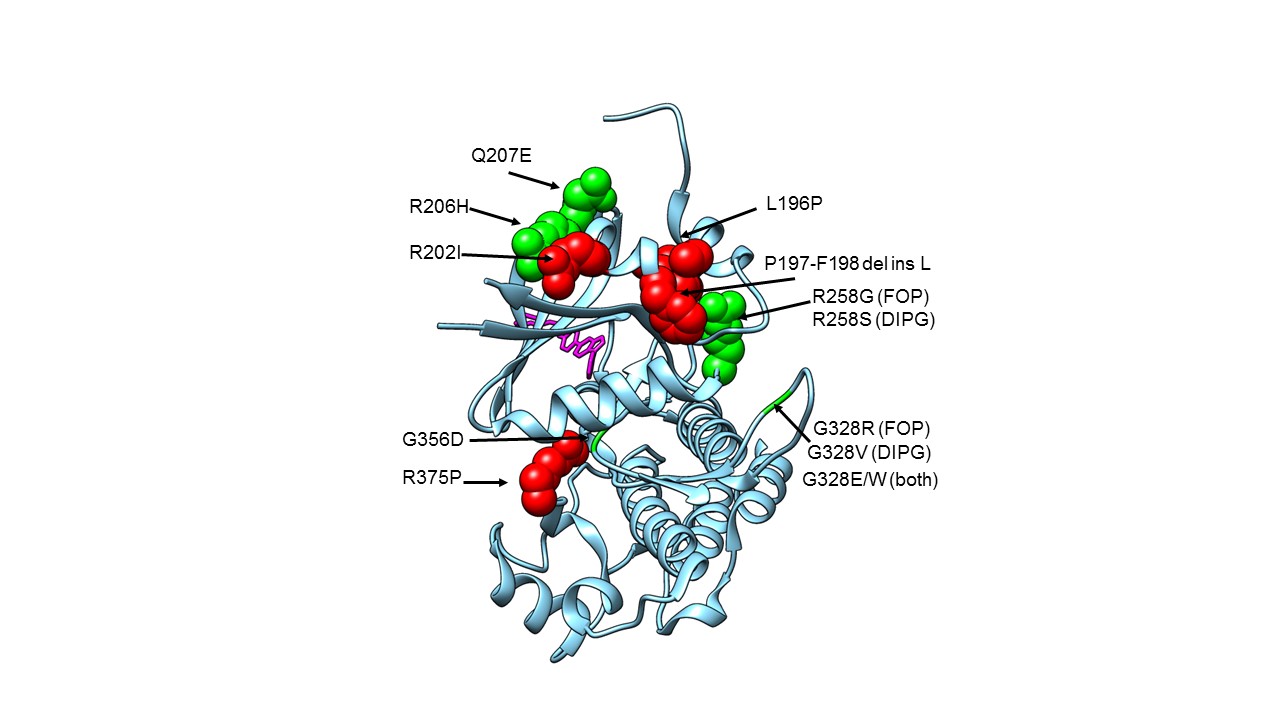
Figure 2. Structure of ACVR1 in complex with dorsomorphin with FOP and DIPG mutation locations shown.
Crystal structure of the ACVR1 kinase domain, with known FOP and DIPG mutation sites shown in space-filling representation (apart from Glycines), and with the dorsomorphin inhibitor shown as sticks in pink. FOP only mutations are shown in red. Mutations found in FOP and DIPG are shown in green. G328V is found in DIPG only. PDB 3H9R
Project overview
The aim of the project is to find some way to inhibit the pathological signalling of the mutant ACVR1 in DIPG. FOP has been studied for longer than DIPG, and the structure of the kinase domain of the protein is known (although we do not have a structure of the active protein). With some of the lessons learnt from FOP, it may be possible to use the known scaffolds of some of the inhibitors of ACVR1 to develop compounds to treat DIPG. The problem with numerous current inhibitors is that many kinases exist in the cell, all with different functions. Many of them have a very similar mode of binding, and thus, finding effective inhibitory drugs that are very selective for a specific kinase is a challenging task. In addition, in DIPG it is necessary to find a drug that can cross the blood-brain barrier.
We will try two crystallographic approaches to search for potential binding compounds. The first will be to test new and existing ATP competitive molecules to improve selectivity for ACVR1. This will be through co-crystallisation of compounds with the enzyme.
The second approach will use a process called fragment screening to find compounds that bind allosteric sites on the protein, and may be able to be built into effective, selective drugs. Fragment screening is a method that x-ray crystallographers use to screen large numbers of compound fragments against a protein to check where and how they bind. In order to see this, the crystallographer has to solve the structure of the protein. This is done by forming crystals from concentrated solutions of protein (not a trivial task), and then soaking them in different compounds that we hope will bind to the protein. The crystals are then placed in a x-ray beam, which has a very small wavelength, such that it is able to resolve atomic distances, and an idiosyncratic diffraction pattern is obtained. This is used to computationally solve the structure of the protein, with any bound molecules. This way we can gradually build up a picture of the shapes of the molecules that can bind to active sites on the protein, and adjust them to bind tighter, or to be more specific for a particular kinase. As an adjunct to this, biochemical and biophysical assays can be done to check the affinity and activity of the compounds.
I have already done an initial small set of fragment screens, and am currently in the process of solving the structures to find bound molecules. Watch this space!
References
Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma
2 Replies to “ACVR1 – the link between FOP and DIPG”