Preparation of CaMKK2 analogs
Pursuant to our aim of preparing a library of small molecule chemical probes around our most promising core scaffold, furopyridine, we sought to develop different chemistries that would allow us to achieve this objective.
In the original synthetic route, 5-bromofuro[2,3-b]pyridine, 1, was able to undergo the Suzuki cross-coupling reaction to install an aryl derivative in compound 2 (Scheme 1). This was followed by bromination on the furan ring at the 3-position, which allows the installation of the crucial cyclopentylbenzoic acid derivative.
Scheme 1: Original Synthetic Route to Furopyridine Analogs
Ar = variety of aryl groups
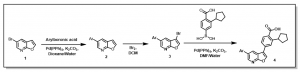
This method led to the preparation of potent CaMKK2 inhibitors. Examples of analogs prepared and their CaMKK2 activities are provided in Figure 1.
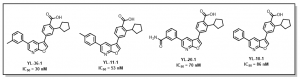
Figure 1: Examples of Previously Prepared Furopyridine Analogs at SGC-UNC
However, the synthetic route, Scheme 2, required repeating step one (1) for each analog prepared, and the bromination step (step 2) limited diversity on the pendant aryl, as many groups get brominated. Figure 2, shows the unintended bromination spot (arrow) on the isoxazole ring, for example.
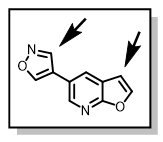
Figure 2: Multiple brominations
Thus, we envisioned that installing the cyclopentylbenzoic acid derivative early in the synthesis (prior to diversification on the pyridine ring), will not only address the bromination issues, but also contribute to preparation of as many analogs as possible within a short space of time. To achieve this, we sought to prepare 3,5-di-bromofuro[2,3-b]pyridine, 5, 5-bromo-3-iodo[2,3-b]pyridine, 6, or 5-bromofuro[2,3-b]pyridin-3-yl-trifluoromethanesulfonate, 7, compounds, Figure 3, with a view to incorporating the cyclopentylbenzoic acid derivative chemoselectively at the 3-position on the furan ring.
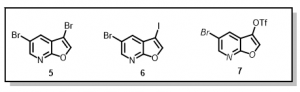
Figure 3: Proposed Furopyridine Scaffolds for Selective Suzuki Reactions
The di-bromo compound, 5, has been achieved but selectivity on the furan ring is poor and we are continuing to review different reaction conditions (Suzuki cross-coupling) to achieve the desired selectivity. Iodination at the 3-position in compound 6 has proven much more difficult than anticipated. In an attempt to achieve the triflate derivative in compound 7, the following synthetic route has been devised, Scheme 2.
Scheme 2: Synthesis of 5-bromofuro[2,3-b]pyridin-3-yl-trifluoromethanesulfonate
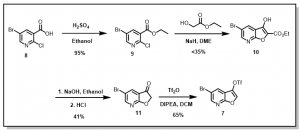
The intermediate ethyl substituted benzoate, 9, was easily obtained in good yield via reaction of the acid, 8, with ethanol in the presence of conc. sulfuric acid (RSC Adv., 2018, 8, 6306, WO 2013/014587). This compound, 9, was further reacted with ethyl glycolate in the presence of NaH to yield the the bicyclic ester, 10, (WO 2013/014587) which was refluxed in aqueous NaOH followed by with aqueous HCl to give the key intermediate furopyridinone, 11, (J. Heterocyclic Chem., 1986, 23, 1465). The compound thus obtained was then converted to the corresponding triflate, 7, followed by chemoselective palladium-catalyzed coupling with select aryl boronic acids (Synlett 2002, No. 3, 501-503, WO 2010/034671), Figure 4.
Although, steps 2 and 3 (Scheme 2) recorded very low yields, gratifyingly we have been able to achieve selectivity for the triflate over the the bromine, with the bromo-derivatives being the only isolable product in most cases, Figure 4.
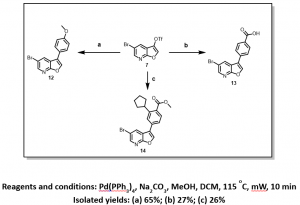
Figure 4: Selective Suzuki Cross-Coupling Reaction
Furopyridine analog via this new synthetic route has been achieved, Scheme 3, and work to create a library of small molecule chemical probes with the promising furopyridine core scaffold is underway.
Scheme 3: Synthesis of 2-cyclopentyl-4-(5-(6-(
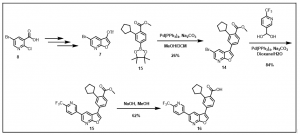
Work towards optimizing the yields in the synthetic route described in Scheme 2 is ongoing. Detailed analogs and their biological data would be provided as they become available.
An elegant solution to the synthesis of the chemoselective intermediate 7. Nice work!