At SGC we are interested in developing protein kinase inhibitors with a high selectivity towards other protein kinases. Based on lead compounds from the literature and our group new and selective CDKL2 inhibitors should be designed, synthesized and tested. Carla Alamillo, a former member of our group, worked on this topic and mentioned in her last entry in the open notebook about this topic (https://openlabnotebooks.org/sns-032-analogues-update/) the pro-mising role of SNS-032 as a starting point for new protein kinase inhibitors (Figure 1).
SNS-032 shows a low Kdvalue for CDKL2 (48 nM) but also for many other kinases. This means the selectivity of this compound towards other protein kinases is low. In contrast, CAF-181 synthesized by Carla (compound 11 in Carla’s entry) shows low Kdvalues for CDKL2 and CDK7 and a high selectivity towards the other kinases tested. Additionally, CAF-181 reveals a moderate CDKL2 IC50value of 417 nM. Therefore, CAF-181 represents a good lead compound for the development of new CDKL2 inhibitors with improved selectivity towards CDK7.
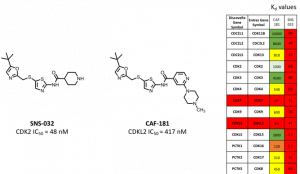
Figure 1. Lead compounds SNS-032 and CAF-181 and their respective Kd values.
To design new compounds the molecular modeling software FlareTM(Cresset®) was used. The first step for the in silico docking is the selection of the crystal structure of the respective protein. Two crystal structures of CDKL2 are available on the PDB website (https://www.rcsb.org) but not published in a journal yet. One crystal structure is co-crystallized with the compound TCS 2312 (PDB-ID 4BBM) and the other with the compound DKI (PDB-ID 4AAA) (Figure 2). The 2D interaction maps (https://www.rcsb.org/structure/4AAA and https://www.rcsb.org/structure/4BBM) have to be flipped vertically to match the same amino acid residues. Interestingly, Val83 (Figure 2, blue box) seems to be an impor-tant interaction partner for the ligands since both compounds reveal two H-bond interactions with Val83.
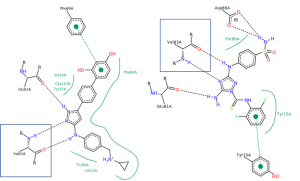
Figure 2. Crystal structures of CDKL2: co-crystallized with TCS 2312 (A, PDB-ID 4BBM, DOI: 10.2210/pdb4BBM/pdb) and co-crystallized with DKI (B, PDB-ID 4AAA, DOI: 10.2210/pdb4AAA/pdb). Black dashed lines indicate hydrogen bonds, salt bridges, and metal interactions. Green solid lines show hydrophobic interactions and green dashed lines show π-π and π-cation interactions (https://www.rcsb.org/structure/4BBM and https://www.rcsb.org/structure/4AAA).
During the in silico docking studies the crystal structure 4BBM was used because the structure 4AAA reveals an unresolved amino acid sequence ending with Ala148 and NME148A (Figure 3B, black box). Unfortunately, this sequence part also contains the amino acid Phe147 that forms a part of the active site and likely interacts with inhibitors. However, the protein structure used for the docking should contain a fully resolved amino acid sequence in this area. The protein structure 4BBM provides a fully resolved amino acid sequence which makes it suitable for in silico docking.
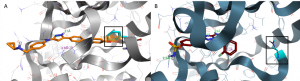
Figure 3. Structure of CDKL2 co-crystallized with either TCS 2312 (orange, A, PDB-ID 4BBM) or DKI (dark red, B, PDB-ID 4AAA). The dashed green and orange lines indicate H-bond interactions and the purple lines CH-π interactions. Docked ligands are shown as sticks. Oxygen and nitrogen atoms are colored in red and blue, respectively.
However, Phe147 in structure 4BBM is not clarified. Instead of the phenyl ring a methyl group is visible. Capping by the docking program FlareTM did not result in phenylalanine but just in a partially aromatic phenyl ring of this amino acid (Figure 3A, black box). This problem was solved by mutation of this position into a phenylalanine.
To investigate the selectivity of newly designed compounds they should also be investigated during the in silico docking into the crystal structure of CDK7 with the PDB-ID 1UA2.
The next step of in silico docking into the CDKL2 structure 4BBM included the validation of the crystal structure and the docking protocol. After preparation and minimization of the crystal structure the co-crystallized ligand TCS 2312 should be redocked into the active site. However, the co-crystallized ligand TCS 2312 shows different bond orders/tautomeric states as well as a different protonation state on the basic nitrogen than the literature compound TCS 2312. To calculate the RMSD value between the binding mode of the structurally corrected and redocked TCS 2312 and the co-crystallized ligand the co-crystallized binding mode had to be corrected because FlareTM can only calculate a RMSD value between structurally identical compounds. Therefore, the co-crystallized ligand was corrected and only the resorcinol moiety was minimized to achieve a planar aromatic system. In this way the binding mode of the co-crystallized ligand was changed as little as possible. Then, the corrected, co-crystallized ligand TCS 2312 was minimized and the structure at physiological pH was generated. Afterwards, this compound was docked into the crystal structure 4BBM. Figure 4A shows the similar orientation of corrected, co-crystallized TCS 2312 (yellow) and the redocked ligand (purple). The similar orientation of both binding modes and the hereby resulting low RMSD value
(1.65 Å) suggest that the corrected CDKL2 protein crystal structure 4BBM and the docking protocol are suitable for the in silico docking of new compounds.
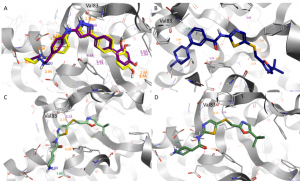
Figure 4. Predicted binding modes of TCS 2312 (A, purple), CAF-181 (B) and SNS-032 (C and D) obtained by docking into the CDKL2 crystal structure with the PDB-ID 4BBM.Binding mode of the corrected, co-crystallized ligand TCS 2312 (A, yellow). The dashed green and orange lines indicate H-bond interactions and the purple lines CH-π interactions. Docked ligands are shown as sticks. Oxygen and nitrogen atoms are colored in red and blue, respectively.
In the Figures 4B-D the docking results of CAF-181 and SNS-032 are shown. The tert-butyl group of both compounds occupies the inner part of the active site whereas the piperazine ring of CAF-181 and the piperidine of SNS 032 point towards the solvent accessible area. The N-atom of the SNS-032 piperidine ring is part of an H-bond interaction with one of the carboxylic acid moieties of either Glu130 (C) or Asp89 (D). Therefore, it might be promising to introduce amide substituents that can interact with those acidic amino acids.
CAF compounds, synthesized and tested previously in our group, were docked into the CDKL2 crystal structure 4BBM and in the CDK7 crystal structure 1UA2. The respective docking scores and electrostatic complementarityTM scores of the resulting docking poses reveal a good correlation to the Kd values of those compounds regarding CDKL2. However, no clear correlation was found regarding docking of CAF compounds in CDK7. This observation could be explained with the larger active site of CDK7 and its many solvent accessible areas that could facilitate binding of structurally diverse compounds.
Interestingly, in silico docking of SNS-032 and CAF-181 in the active site of CDK7 reveal curved orientations of both compounds (Figure 5). The green color of the electrostatic complementarityTM surface of the SNS-032 binding mode in Figure 5C indicates a high electrostatic complementarityTM of the ligand to this active site which could be the reason for the high inhibitory activity of SNS-032 regarding CDK7.
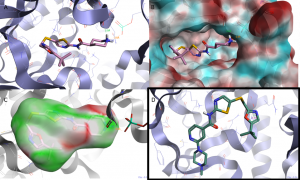
Figure 5. Predicted binding modes obtained by docking into the CDK7 crystal structure with the PDB-ID 1UA2:
(A) SNS-032. (B) SNS-032 with the electrostatic potential surface of CDK7. (C) SNS-032 with its electrostatic complementarityTM surface. (D) CAF-181. The dashed green and orange lines indicate H-bond interactions and the purple lines CH-π interactions. Docked ligands are shown as sticks. Oxygen and nitrogen atoms are colored in red and blue, respectively.
Based on this observation new CDKL2 inhibitors should reveal restrained flexibility to avoid the curved orientation in CDK7 and thereby increase the selectivity towards CDK7. Among others this could be achieved by replacement of the flexible thio-methylene linker by a more rigid linker. In my next entry I will show the docking results of more rigid compounds obtained by both in silico docking into the crystal structure of CDKL2 and CDK7.
Please feel free to suggest other ideas for CDKL2 inhibitors with increased selectivity towards CDK7. If you have any questions or comments, please let me know.