Introduction:
The outbreak of COVID-19 demonstrated the scarcity of drugs in the development pipeline to decrease the mortality and morbidity caused by the new pathogenic SARS-CoV-2 virus. Despite the improved diagnosis and screening processes, broad directly-acting anticoronavirus drugs were not immediately available to treat viral infections. In response to this health threat, the Rapidly Emerging Antiviral Drug Discovery Initiative – Antiviral Drug Discovery Center (READDI-AC) was established to development of a pipeline of oral, direct-acting antiviral hits, leads, and drug candidates with broad activity against alphaviruses, flaviviruses, coronaviruses, and filoviruses with high pandemic potential. Importantly, READDI-AC currently focuses on highly conserved viral RNA polymerase (RdRp), helicase (Hel), and proteases (Pro) that are essential for positive and/or negative strand emerging virus replication and pathogenesis.
Helicase inhibitors as antiviral strategy:
DNA and RNA helicases are critical components of the viral replication-transcription complex. The discovery of Pritelivir demonstrated that inhibitors of the herpes simplex virus DNA helicase-primase could block the replication of strains that had developed resistance to the polymerase inhibitor acyclovir (Birkmann et al., 2022). SAR-CoV-2 RNA helicase (NSP13) catalyzes the separation of duplex oligonucleotides into single strands in a nucleotide triphosphate (NTP) hydrolysis-dependent manner. Due to high sequence conservation across the coronaviruses and its essential role in viral replication, NSP13 has been identified as an attractive drug target (Yazdani et al., 2021). Newman et al. recently reported 65 fragment hits of NSP13 by crystal soaking, which identified several druggable pockets on the helicase.
DNA and RNA helicases contain two conserved RecA-like domains that have been shown to be druggable. Therefore, the synthesis of small arrays based on known chemotypes that target RecA-like domains is a targeted approach to developing new helicase inhibitors. The HSV DNA helicase and CoV RNA helicases belong to the same SF1 subfamily. This blog post describes our chemistry campaign around the sulfonamide chemotype that is found in the HSV DNA helicase inhibitor Pritelivir (Kleymann, ACC 2004) (Fig 1). Although the crystal structure of Pritelivir with HSV helicase has not been solved yet, the allosteric binding site on the RecA domain has been identified by mutagenesis (Birkmann et al., 2022).
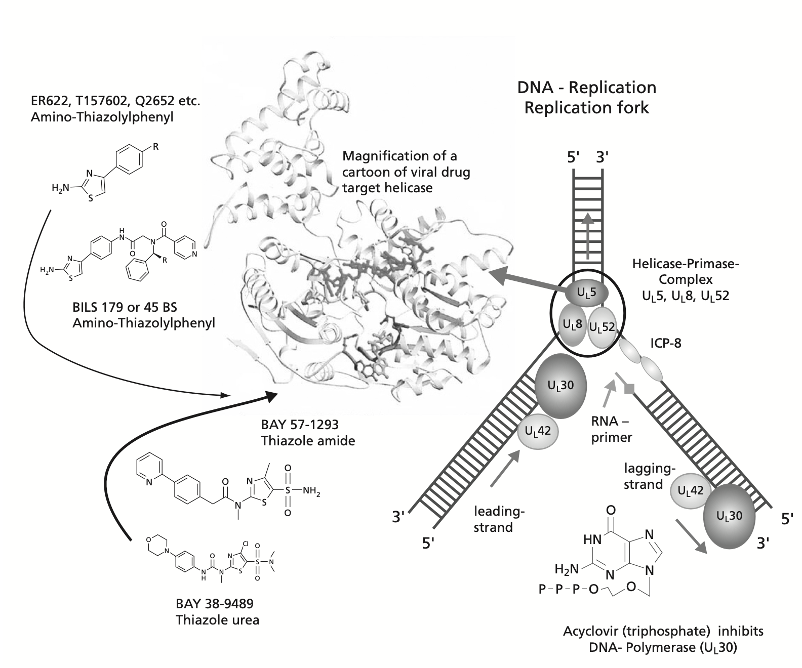
Figure 1: HSV Helicase inhibitors (Kleymann, 2004)
Chemistry Campaign:
In the early 2000s, multiple laboratories reported the discovery of thiazoles and thiazolylamide chemotypes that were active against HSV in phenotypic assays. Optimization of these compounds into clinical candidates coincided with the identification of HSV helicase-primase as their molecular target (Fig 1). The binding site of Bay 57-1293 (Pritelivir) was identified by mutagenesis to lie on the interface of the two RecA-like domains. To design analogs of the core sulfonamide chemotype found in Pritelivir for screening on SF1 helicases, we deployed the following chemistry strategies:
A: Structural modification of Pritelivir
- N-alkyl chain: N-alkyl substitution of Pritelivir was optimized from the early sulfonamide inhibitor for inhibition of HSV helicase. Therefore, we removed the alkyl substation at the 2-amine of thiazole to increase the increase the likelihood of cross-helicase activity.
- The biphenyl moiety: Biphenyl moiety showed an increased potency for HSV helicase (Kleymann, 2004). In our library, we avoided the incorporation of biphenyl-like substituents (Fig 2)
- Thiazole sulfonamide: Thiazole sulfonamide and thiazoles were preferred chemotypes for HSV helicase inhibition. For the helicase-targeted array, five different 5-membered rings with diverse functional groups were synthesized.
B: Library synthesis consideration
- Avoided lengthy multistep linear synthesis
- Allowed mix and match of the thiazole and phenyl scaffolds
- Used calculated physicochemical properties to target drug-like chemical space
- Designed two initial 10 x 5 arrays for a total of 100 analogs
C: Data-driven further optimization
- Screening against SARS-CoV-2 nsp13 helicase was performed by SPR and ATPase assays. Compounds were also assessed in a SARS-CoV-2 viral replication assay.
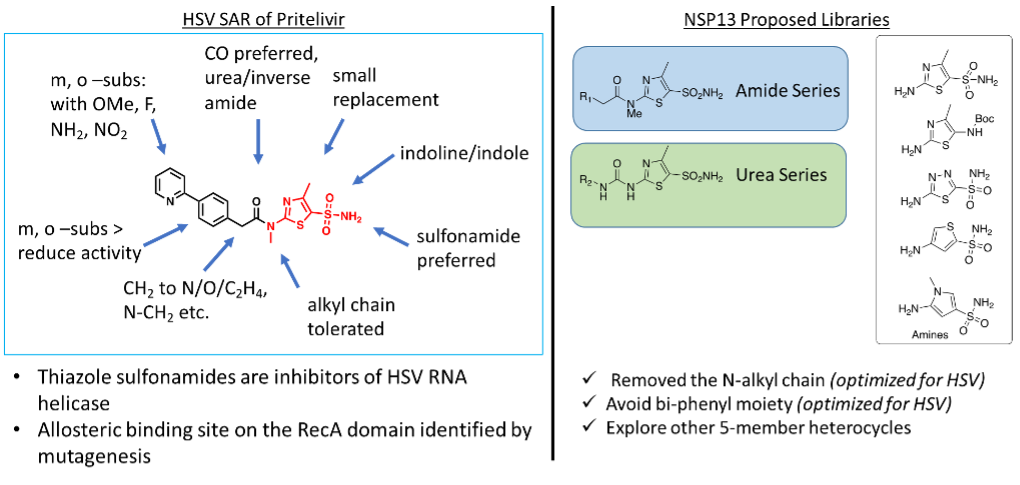
Figure 2. NSP13 chemistry campaign based on HSV helicase Pritelivir. R1 and R2 = Biphenyl for controlled analog; heteroaromatic, fused-ring system, phenyl, benzyl, substituted alkyl chain, etc.
General synthesis scheme:
Thiazole series contains two series of compounds, amide and urea. The following general schemes are used to access the designed library:
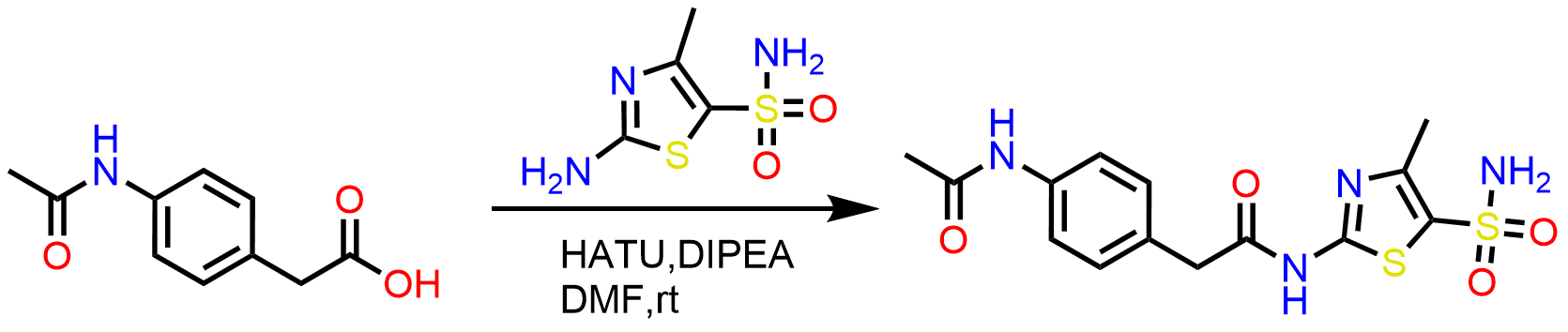
Scheme 1: Synthesis of amide series using standard amide coupling from the commercial building blocks.
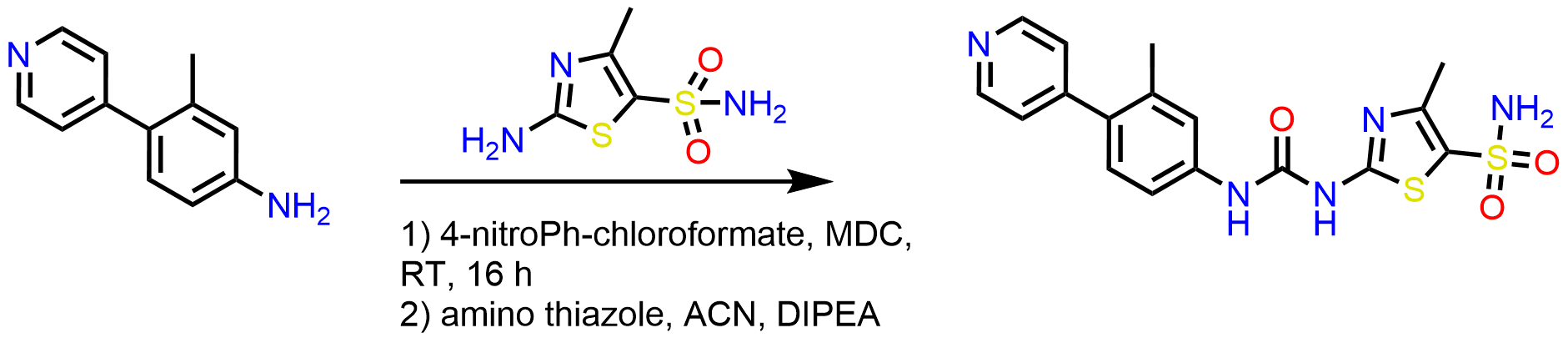
Scheme 2: Synthesis of urea series using CDI or 4-nitrophenyl chloroformate to activate the amine in the presence of MDC, room temperature for 16 h, followed by adding amino thiazole in acetonitrile.
In-Vitro HTS screening:
To date, 40 new thiazole analogs were synthesized as outlined in Schemes 1 and 2. 25 of these analogs have been screened against SARS-CoV-2 nsp13 by in-vitro ATPase and SPR assays (Figures 3 to 5).
As expected, the HSV helicase inhibitor Pritelivir did not show any cross-activity in the NSP13 ATPase assay. However, in the preliminary ATPase assay, three of the sulfonamide analogs showed moderate activity ranging, with IC50s ranging between 8 to 57 μM.

Figure 3. NSP13 ATPase assay of Pritelivir and its representative analogs

Figure 4. Representative structures and ATPase IC50 of selective scaffolds
Physicochemical Properties of synthesized analogs:
Aqueous solubility is one of the important drug properties of a tractable lead compound. Lipophilicity (cLogP) and melting point have been shown to be good predictors of aqueous solubility (Tinworth et al. 2020). The synthesized sulfinamide analogs have cLogP < 3 (Fig. 5), which is comparable to Pritelivir (cLogP = 2.78, MP °C = 177). All the labeled shapes in figure 5 showed some activity either in ATPase or SPR assay against NSP13. The top two compounds, RA-0001250-01 and RA-0001247-01 (ATPase IC50 8 µM and 17 µM, respectively), have cLogP of 1.82 and 0.93 and melting points of 176 °C and 175 °C, respectively. However, these two compounds were not active in the NSP13 SPR assay (Fig 6). At this stage, a secondary assay is required to confirm activity against NSP13 or SARS-CoV-2.
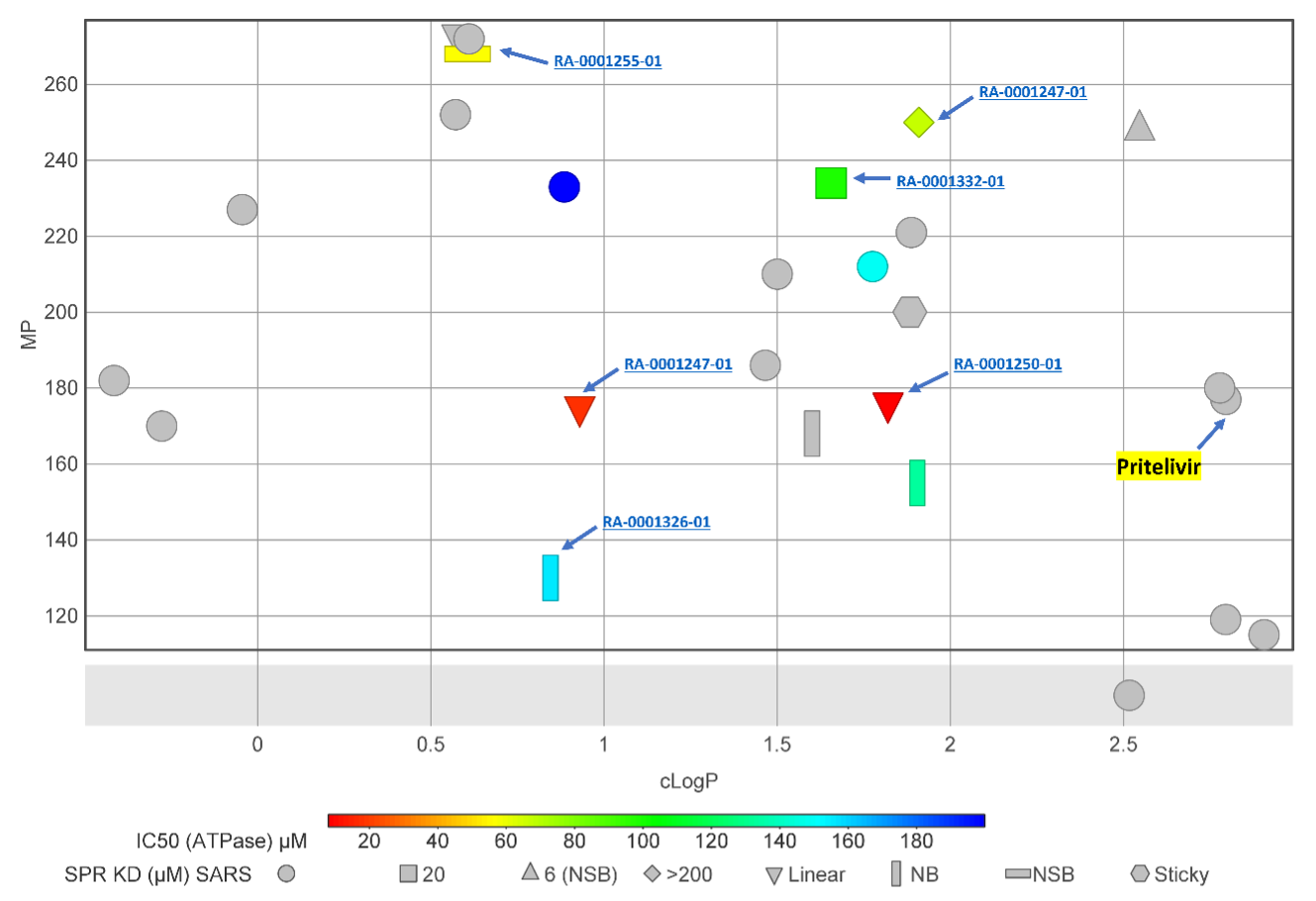
Figure 5. 2D representation of physicochemical properties and in-vitro activity profile of thiazole analogs. NSB = Non-saturable binding, NB = No binding, Sticky = Sticky property in SPR assay
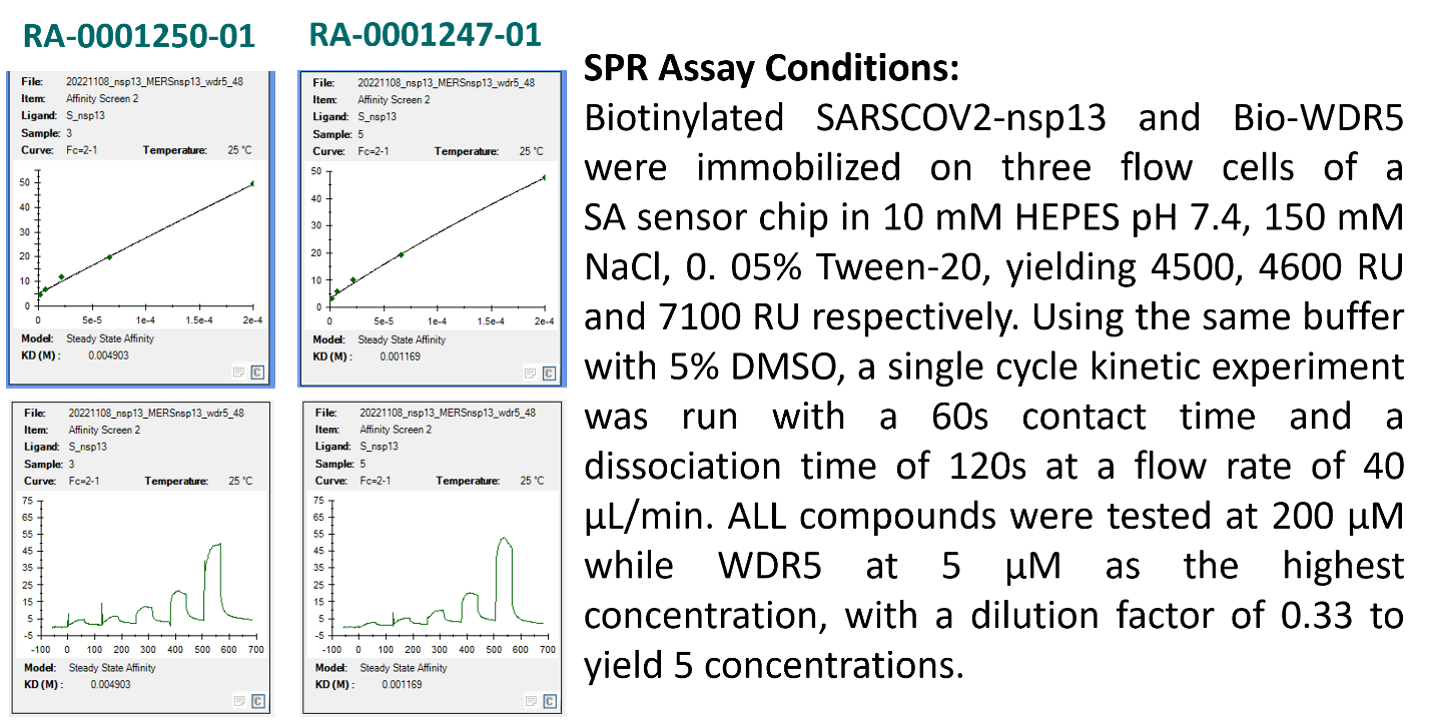
Figure 6. The SPR sensorgram of the top two thiazoles RA-0001250-01 and RA-0001247-01 and the experimental condition
Conclusion:
An efficient 1 or 3 step synthesis of two arrays based on the Pritelivir thiazole sulfonamide chemotype was used to deliver 50 analogs for screening on SF1 helicases. The initial screening data identified three analogs with micromolar activity in the NSP13 ATPase, but activity was not confirmed by the SPR assay. Further work includes the screening of the array in a SARS-CoV-2 cell-based assay as well the development of a model of the thiazole sulfonamide binding site to direct synthesis of additional analogs.
To know more about READDI: https://www.readdi.org/
Interested in participating in AViDD Open Science? Check here
References:
Birkmann, A.; Bonsmann, S.; Kropeit, D.; Pfaff, T.; Rangaraju, M.; Sumner, M.; Timmler, B.; Zimmermann, H.; Buschmann, H.; Ruebsamen-Schaeff, H. Discovery, Chemistry, and Preclinical Development of Pritelivir, a Novel Treatment Option for Acyclovir-Resistant Herpes Simplex Virus Infections. Journal of medicinal chemistry 2022, 65 (20), 13614-13628. DOI: 10.1021/acs.jmedchem.2c00668
Yazdani, S.; De Maio, N.; Ding, Y.; Shahani, V.; Goldman, N.; Schapira, M. Genetic Variability of the SARS-CoV-2 Pocketome. Journal of Proteome Research 2021, 20 (8), 4212-4215. DOI: 10.1021/acs.jproteome.1c00206
Newman, J. A.; Douangamath, A.; Yadzani, S.; Yosaatmadja, Y.; Aimon, A.; Brandao-Neto, J.; Dunnett, L.; Gorrie-Stone, T.; Skyner, R.; Fearon, D.; et al. Structure, mechanism and crystallographic fragment screening of the SARS-CoV-2 NSP13 helicase. Nat Commun 2021, 12 (1), 4848. DOI: 10.1038/s41467-021-25166-6
Kleymann, G. Helicase primase: targeting the Achilles heel of herpes simplex viruses. Antivir Chem Chemother 2004, 15 (3), 135-140. DOI: 10.1177/095632020401500303
Kleymann, G. Discovery, SAR and medicinal chemistry of herpesvirus helicase primase inhibitors. Current Medicinal Chemistry-Anti-Infective Agents 2004, 3 (1), 69-83. DOI: 10.2174/1568012043354242
Tinworth, C. P.; Young, R. J. Facts, Patterns, and Principles in Drug Discovery: Appraising the Rule of 5 with Measured Physicochemical Data. Journal of medicinal chemistry 2020, 63 (18), 10091-10108. DOI: 10.1021/acs.jmedchem.9b01596
Anwar, beautiful results (and beautiful blog post)! Could it be that the ATP-ase hits are not binding in the SPR assay because the enzyme is locked in an incompatible conformational state?
We don’t know exactly why the discrepancy, but it is a good point to figure it out. Thank you very much for your comments. We will keep you posted!
Anwar
Permanent solution for Herpes___________________________________Email Robinsonb uc ler { @gmail } com…………
Hi,
Do you mind clarifying your comment?
Thank you.
Anwar