By Zachary Davis-Gilbert, Anwar Hossain, and David H. Drewry*
In conjunction with our efforts to target TBXT via non-covalent small molecules, we have also been developing covalent compounds that bind to CYS122, which has previously been shown to be accessible by afatinib (pdb code 6ZU8). In this blog post, I will discuss the progress made on improving the binding affinity of new covalent compounds targeting CYS122 as well as some of the directions we are taking to further improve binding affinity to TBXT and reduce binding affinity towards EGFR. The synthetic work has been primarily driven by Zach and our CRO – Piramal, and mass spec labeling and crystallographic data have been performed by Joseph Newman and Nergis Imprachim at Oxford. This blog will be two parts and will also contain a Zenodo (https://doi.org/10.5281/zenodo.6629195) release of all compounds we have tested and their % labeling of brachyury.
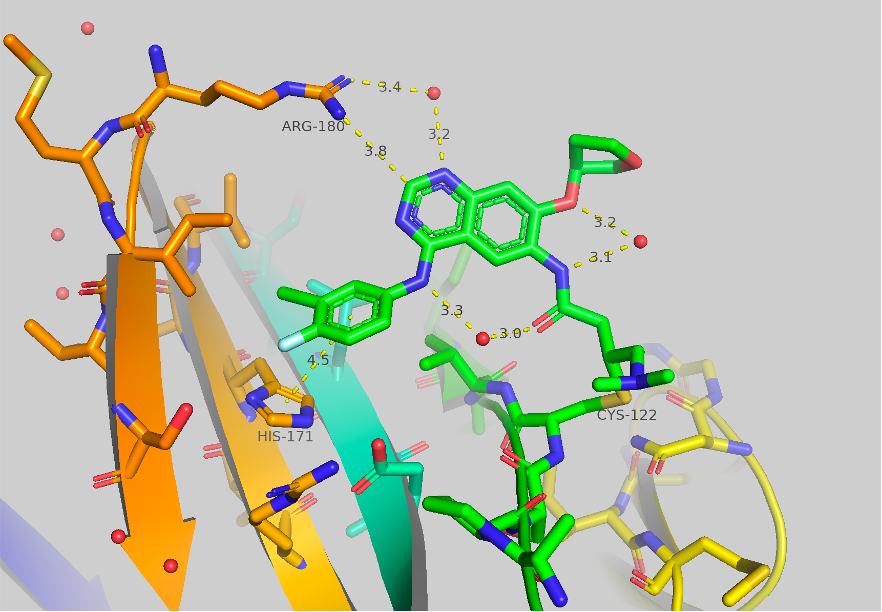
Figure 1 Crystal structure of afatinib bound to TBXT, 6ZU8. Contacts to HIS171 and ARG180 are shown as well as some potentially important bridged water molecules.
The crystal structure of afatinib bound to TBXT is shown in Figure 1. Contacts to HIS171 and ARG180 are shown, although they are quite long (4.5 & 3.8 Å). A few potentially significant water molecules are present between the amide link to the covalent warhead and the ortho-ether and the NH aniline. A water molecule is also shown between ARG180 (3.4 Å) and the 1-N of the quinazoline core (3.2 Å). We initially aimed to engage ARG180 and to improve the pi-pi interaction of HIS171.

Figure 2 Cores for covalent inhibitor SAR. Left: Benzamide bioisosteres. Right: Quinazoline core focusing on SAR of the aniline in the 4 position.
For this work, we developed two directions for SAR. The first direction we decided to take was to “deconstruct” the quinazoline ring to the corresponding benzamide bioisostere. This route would allow SAR divergence to occur at the final step using amide coupling from the corresponding benzoic acid. We also hypothesized that this would help us move away from potential overlap with EGFR inhibition as it represents a drastic modification of the EGFR hinge binding moiety of afatinib. The second method would be to continue working with the quinazoline core from afatinib (using methoxy as a simpler substituted ortho-ether) this route would be slower as each aniline would need to start from the parent 4-chloro-7-fluoro-6-nitroquinazoline (later a faster route was found but has not been implemented by our team yet).

Figure 3 afatinib and best compounds from initial screening of new anilines
After preparing the parent benzoic acids we screened an array of anilines and measured their affinity towards brachyury by mass spec % labeling, with a protein concentration of 5 mM in buffer. Shown in Figure 3 are the two initial top hits for the meta and para benzoic acids and afatinib as reference. The best compound found for the meta-analog was the parent 4-chloro-3-fluoro aniline (the direct bioisostere analog to afatinib), no further improvements were found in this series. To our delight, a new compound from the series with the amide linkage para to the electrophilic covalent warhead was found which showed improved affinity towards TBXT at both 50 and 5 mM concentrations. This compound contained a 3-N,N-diethylsulfonamide in place of the parent fluoro group. This compound was labeled at roughly 3-fold better than afatinib at 5 mM in 1 h. With ZDG-13807 as our new lead compound, we began to examine its SAR to further increase binding.
Figure 4 Highlighted regions of UNC-ZDG-138-7 for SAR
Our plan to examine the SAR of this lead compound is highlighted in Figure 4. These circled positions presented themselves as the synthetically easiest positions to explore. Replacing the dimethylamine (blue circle, Figure 4) on the covalent warhead with different amines only led to a decrease in % labeling. Next, we examined the amine of the sulfonamide piece (maroon circle, Figure 4). Replacing the diethylamine with isopropylethylamine or piperidine both yielded increased potency (51% and 46% labeling 1 h @ 5 mM, respectively). We were able to obtain crystal structures of both compounds.
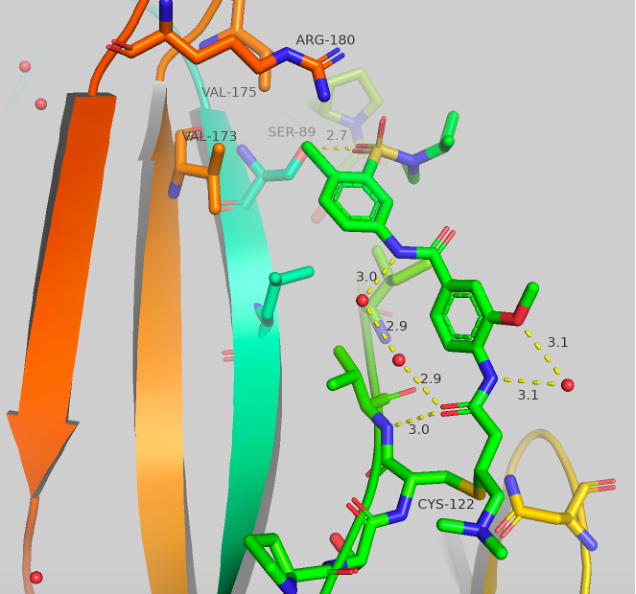
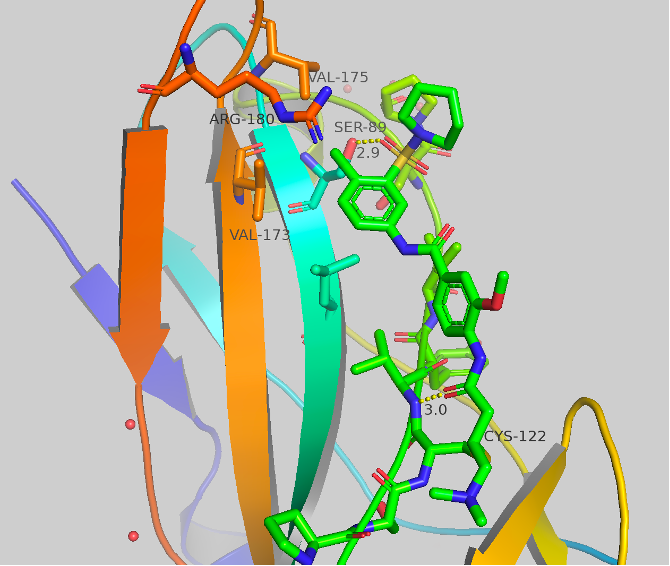
Figure 5 Top: Structure and crystal structure of UNC-ZDG-173-1 Bottom: Structure and crystal structure of CF-8-115
The structures and crystal structures are shown in Figure 5. These compounds make some major directional differences when compared to afatinib (Figure 1). The cysteine residue remains the same, but the aniline portion of the molecule, which previously resided near HIS171, now points towards a hydrophobic region (this “groove” or “pocket” is the same region that is occupied in the A’ series which we have previously blogged). The new major contact formed is between the oxygen of the sulfonamide and SER89 at 2.7-2.9 Å. There orientation of the second oxygen and the dialkylamine varies between the two compounds, but no other significant contacts are made. The chlorine atom now sits sandwiched between VAL173, VAL175 and ARG180. UNC-ZDG-173-1 contains some potentially important water molecules, specifically, a water molecule still resides between the warhead and the methoxy group and a water bridge is visible between the two amides. These water bridges are not present in CF-8-115, but the orientation of the methoxy and amides remain consistent between these two compounds.
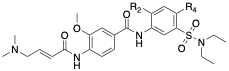

Table 1: SAR of UNC-ZDG-138-7
The final modification we have made that has provided improvements to affinity is the replacement of the chlorine shown in Table 1. We found that this replacement is quite sensitive, (which may be expected due to its proximity to 3 amino acids). For example, replacement of the chlorine with morpholine completely abrogated labeling, potentially due to volume mismatch. Of these changes, replacement of the chlorine functional group with cyclopropyl (13 in table) resulted in 81% labeling under our standard conditions. This represents a roughly 4-fold increase from the parent chlorine and an 8-fold increase in labeling when compared to afatinib and remains our best compound in this series to date.
To conclude this first part of the series, we have made very meaningful progress on a new scaffold targeting the CYS122 on brachyury. The best new compound we have found has an 8-fold increase in labeling affinity compared to afatanib and there appear to be more avenues to explore to continue making improvements. In the next section, I will discuss progress made on the quinazoline core.