Our chordoma research focused on developing small reversible molecules targeting the transcription factor TBXT which codes for the brachyury protein. Brachyury contains several shallow pockets, and through an x-ray crystallography fragment based approach we previously identified several series of small fragments bound to it (TBXT Fragment Hits). Our medicinal chemistry project aimed to grow the initial fragment hits to increase their affinity for brachyury. Several hundred commercial fragments were screened in SPR, and some were identified as potential hits.
We have screened over 3500 small molecules in SPR against TBXT (SPR Raw Data). The SPR assay has been run in screening mode, so each curve needs to be looked at carefully. In addition, repeat runs at lower concentrations combined with screening in orthogonal assay formats will help build confidence in the ligands. A key goal is to deliver high-quality non-covalent probes. Figure 1 lists some key features of a quality probe.

Figure 1. Key Features of Reversible ligand
In the beginning, our synthesized analogs comprised at least ten different series of molecules. We are now working primarily on three series of molecules due to the reproducible activity of analogs, accessible chemistry, ease of diversification, and potential to obtain compounds with appropriate properties. The top three classes of ligands include the thiazole, lactam, and quinazoline scaffolds.
Thiazole Series
In our previous thiazole blog, we explained our strategies and progress for this class of molecules. Here, we are presenting some recent results. As shown in figure 2, the thiazole series interacts with ARG-180 through an H-bond interaction and makes a water bridge to brachyury involving the thiazole N (CSC027898502). Additional water interaction involves the morpholine.
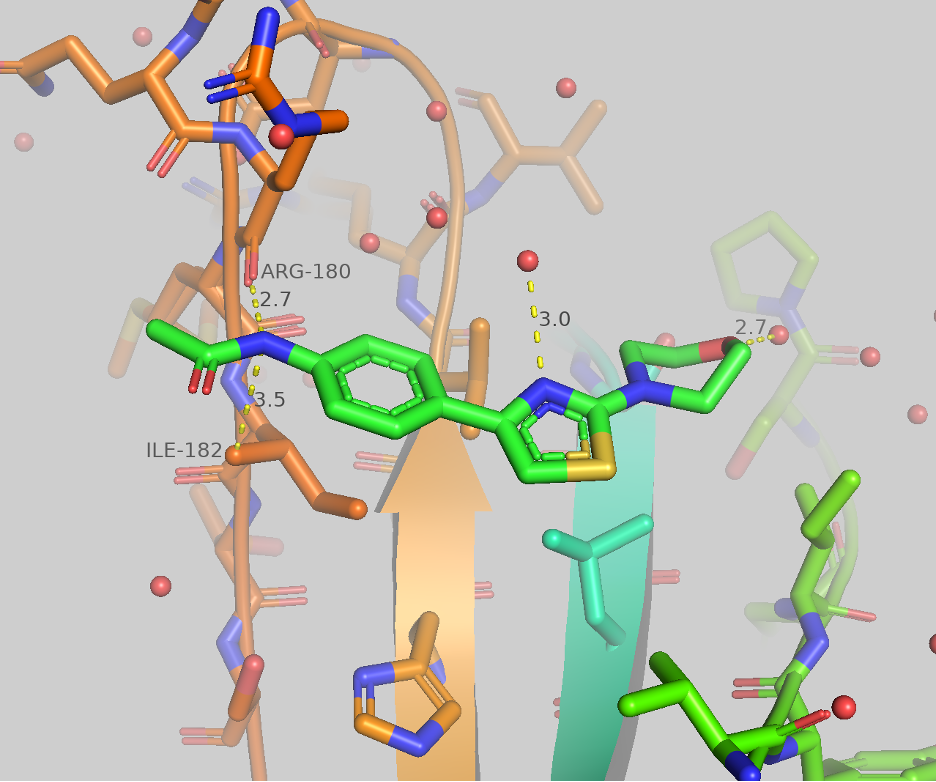
Figure 2: Thiazole CSC027898502 crystal structure at A’ pocket of TBXT (PDB 5QS9).
Initially, we screened a few hundred commercial compounds and a couple of notable hit molecules were identified in the A’ pocket (Figure 3, CSC027898502). We used this as our reference template and made analogs to improve the potency and physicochemical properties. The ref compound CSC had lead-like properties but showed very weak SPR KD (316 μM). In our initial modification approach, we changed acetamide to cyclopropyl-to cyclobutyl- to cyclopentyl moiety. Among these three different substitutions, the presence of the cyclopropyl moiety increased the potency from 316 μM to 19 μM.
In the next round of optimization, we focused on three regions to optimize: the morpholine position, the thiazole core, and the phenyl moiety (Figure 4B). Replacing morpholine with piperazinone improved activity with predicted solubility at the same time. Methyl substitution on the phenyl moiety also provides a good binder. The predicted ADME properties also showed an improved profile compared to the parent hit thiazole (Figure 3).
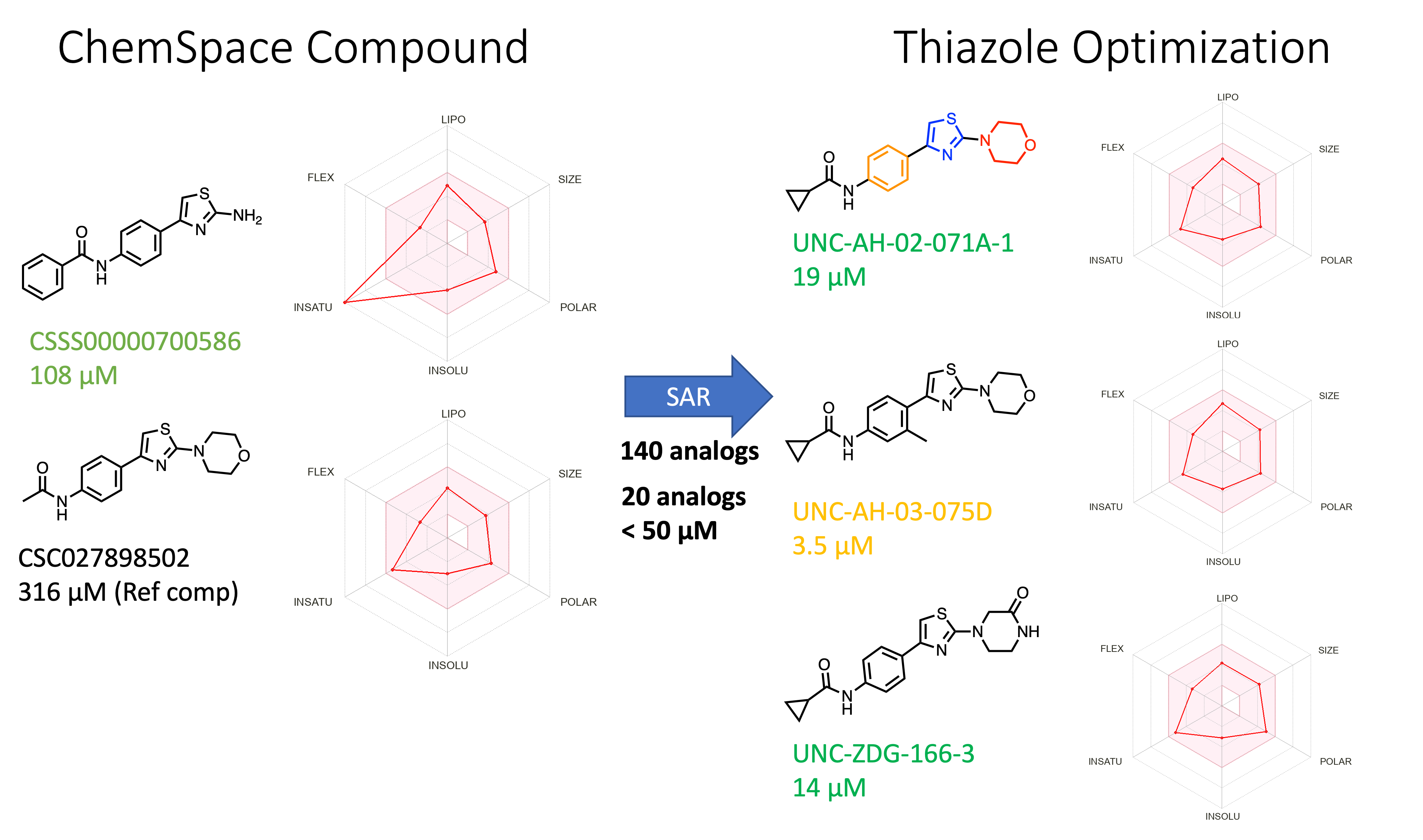
Figure 3. Initial thiazole hit (CSC027898502) and examples from optimization [Color code from manual data analysis and visualization: Green = Excellent Curve Fitting Data; Orange = Good Curve Fitting Data with outlier data point; Red = Not a good curve fitting data; Radar Chart represent predicted ADME properties using SWISSADME]
- SPR activity for multiple examples from the scaffold (20 of 140 analogs < 50 μM)
- Low molecular weight (<350)
- Robust synthetic route (Figure 4)
- Ligand for A’ pocket can be extended to Cysteine residue (CYS122) to develop covalent inhibitors (Link)
- Leadlikeness
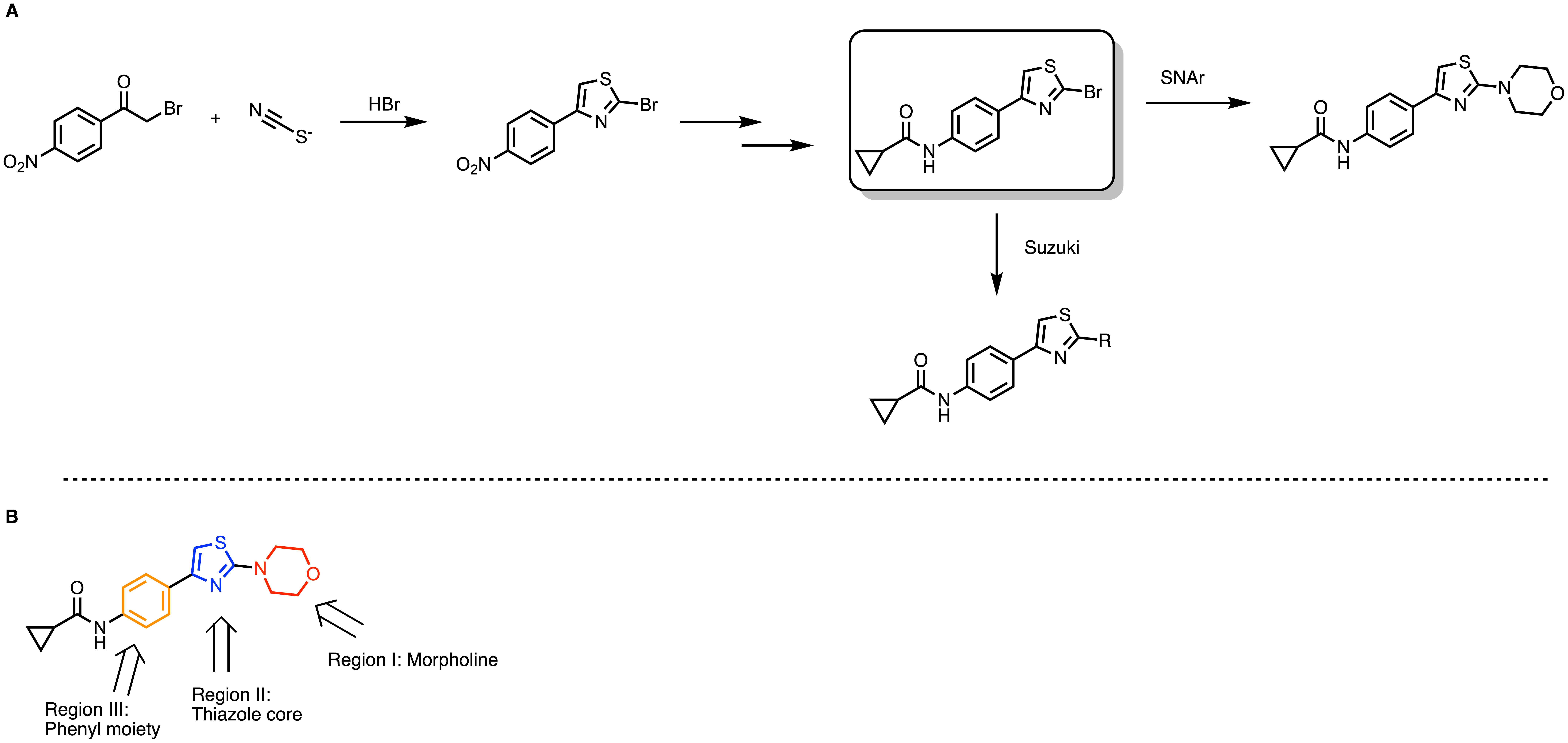
Figure 4. General synthetic scheme of thiazoles (A). SAR strategies for thiazoles (B).
Pyrimidines (replacement of thiazole core)
In addition to thiazoles, we explored the replacement of the thiazole core with a pyrimidine. This change is useful from a synthetic chemistry standpoint (easy to make and modify), and we hypothesized that it would allow for a similar shape and angle between the substituents on the heterocycle (Figure 5). Gratifyingly, this core replacement retains binding to brachyury as measured by SPR. At this moment, we don’t have a crystal structure of a pyrimidine to know the detailed interactions of this molecule with the A’ pocket, but this finding expands opportunities for optimization.
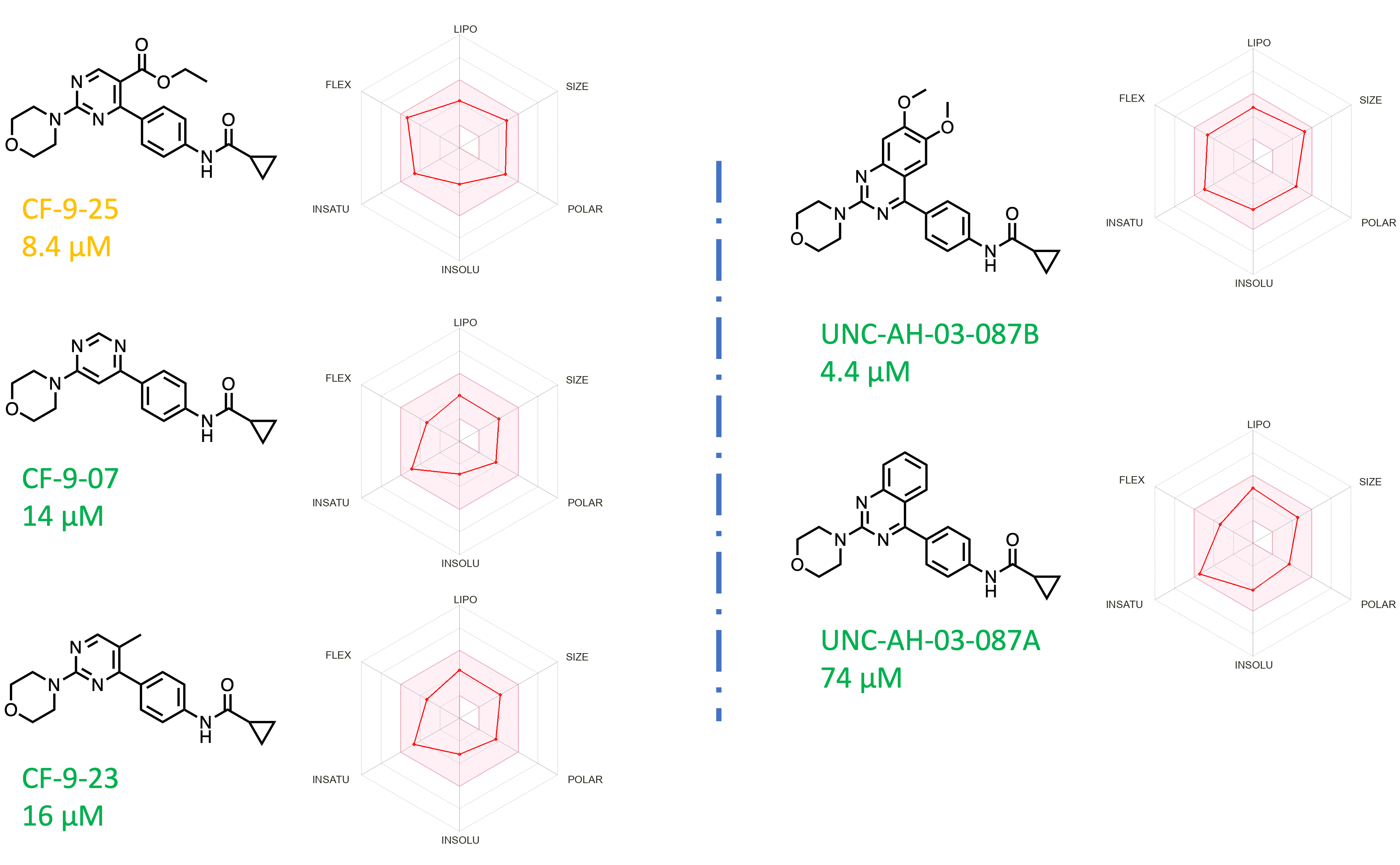
Figure 5. Thiazole core replacement. [Color code from manual data analysis and visualization: Green = Excellent Curve Fitting Data; Orange = Good Curve Fitting Data with outlier data point; Red = Not a good curve fitting data; Radar Chart represent predicted ADME properties using SWISSADME]
We identified the lactam series (previous post) compound from the commercial source analog MolPort -020-049-143 (Figure 6A). The top two lactams showed moderate SPR activity with favorable calculated physicochemical properties. Later, we initiated structure-activity relationship studies and designed and synthesized over 250 analogs (Figure 6B). The synthesis took place at Piramal. Our focus was to modify both the left-hand phenyl and the right-hand aniline (Figure 7B.) We performed a halogen, methyl, and methoxy scan on the left-hand phenyl, suggesting that halogen may be better tolerated than any alkyl substitution. The right side aniline substitution was explored in two different manners: i. introduction of an amide linker with their lactam amine followed by further aryl modification (Figure 7B), ii. Modification of the aniline with various substitutions (For details @Zenodo).
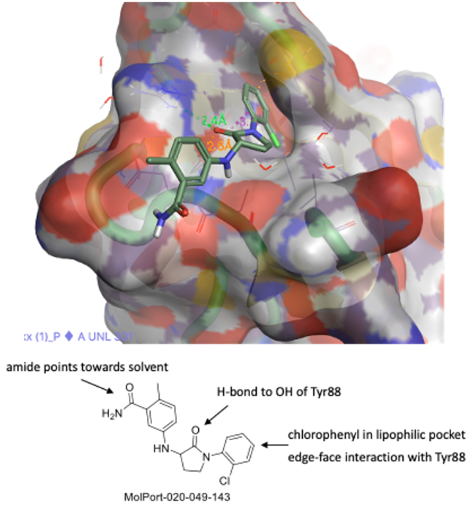
Figure 6A. Lactam hit molecule (MolPort-020-049-143) and its interactions with pocket F (TBXT).
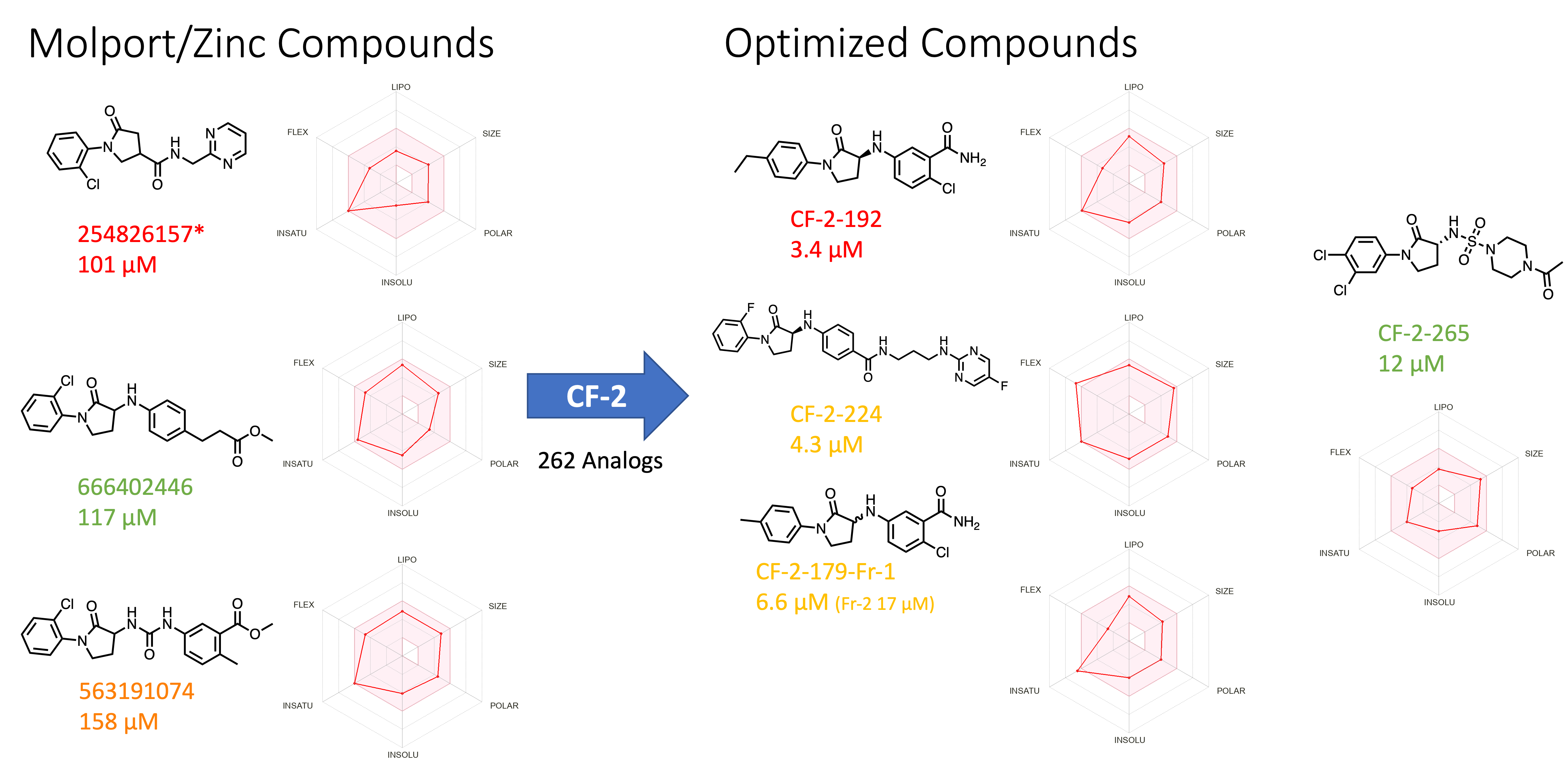
Figure 6B. Lactam series commercial compounds and optimization. [Color code from manual data analysis and visualization: Green = Excellent Curve Fitting Data; Orange = Good Curve Fitting Data with outlier data point; Red = Not a good curve fitting data; Radar Chart represent predicted ADME properties using SWISSADME]
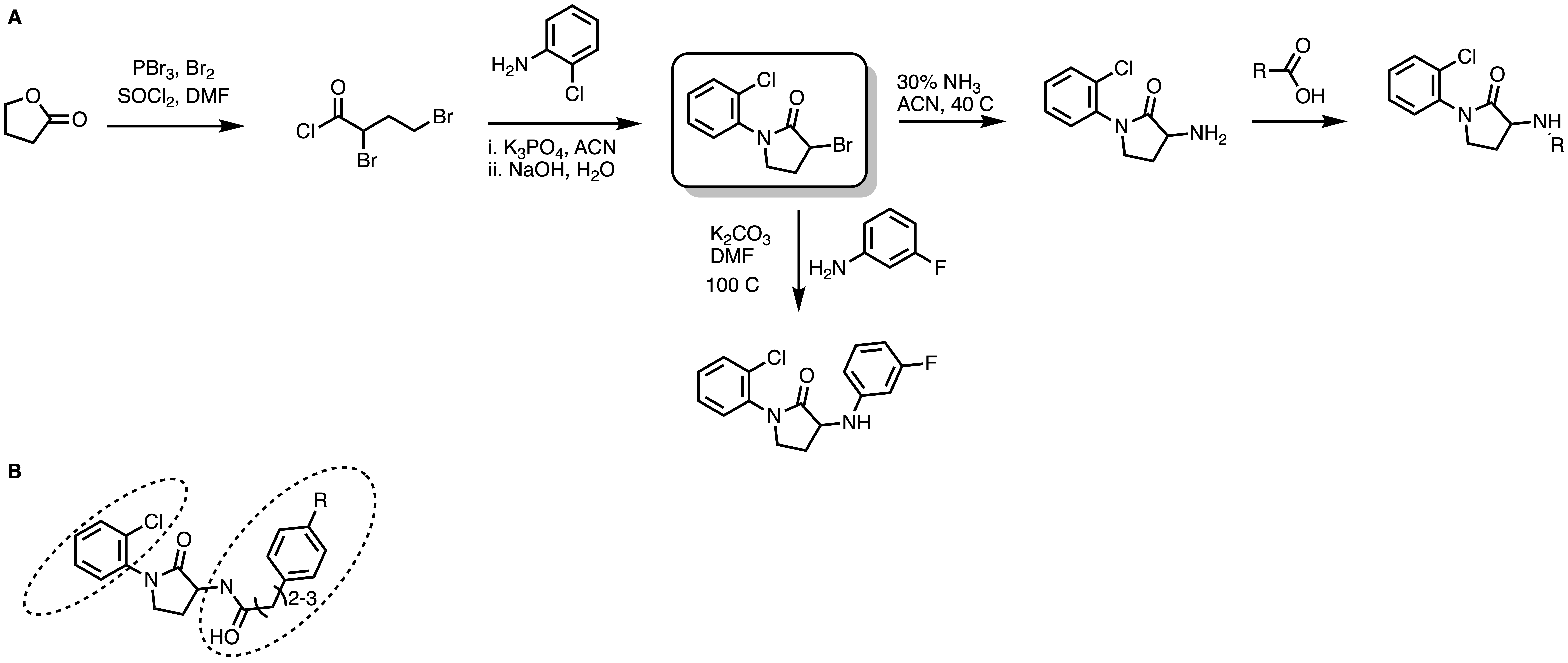
Figure 7. General synthetic scheme for lactams (A). SAR Strategies for lactams (B).
The lactam scaffold showed an interesting 3D conformation and interaction with the target site (Figure 8). In this crystal structure, the carbonyl oxygen of the lactam makes two contacts with TYR-88 (2.5 Å) and ARG-174 (3.1 Å). We believe this interaction fixes the compound in this pocket. One phenyl moiety makes a stacking interaction with MET-181 and the other aniline moiety points towards the solvent front. Based on these crystal interactions, we have space to grow this molecule. We can make additional contact with GLU-41 with appropriate functionalization and introduce some bulkier substitution. Moving forward, we have scope to optimize the series keeping the lactam moiety intact. Others have commented on and reported the potential of lactams as useful cores for medicinal chemistry projects (Saldívar-González et. al.,).
We currently have a few challenges for this series, including the synthesis of compounds in an enantiomerically pure form and synthetic challenges to produce specific modifications efficiently. Our med chem team works on a case basis to overcome these challenges.
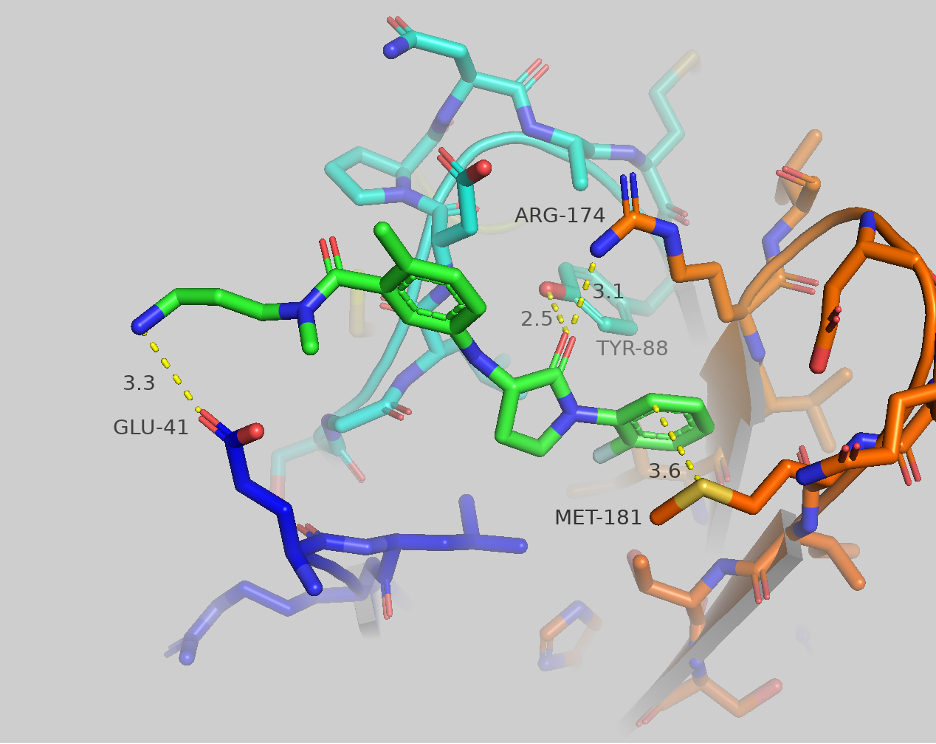
Figure 8. Crystallography of CF-2-173 (lactam).
Key properties of the Lactam series:
- SPR activity for multiple examples from the scaffold (14 of 262 analogs < 50 μM)
- Low molecular weight (<350s)
- Robust synthetic route to access common intermediates (Figure 7A)
- Characteristic 3D space
- Leadlikeness
- Potential for bivalent degrader development
Quinazoline Series
Our third series, quinazolines, consists of analogs that emerged from molecules derived from afatinib. While screening the afatinib-inspired covalent series, we also screened the intermediates made before the installation of the covalent warhead. The rationale behind this is to know the affinity of the non-covalent moiety against the target protein. As shown in figure 9, the non-covalent core showed several key interactions with the pocket of brachyury (Figure 9: representative covalent analog CF-8-41). The core quinazoline moiety is securely positioned in the lipophilic groove. The oxazole moiety makes two contacts with ARG-169, and the phenyl moiety in between the oxazole and the quinazoline moiety makes a pi-pi stacking interaction with HIS-171. These interactions indicate that this series could be a standalone non-covalent TBXT ligand. With this hypothesis, we screened a small set of molecules with diversification at the aniline moiety, and a significant number showed excellent SPR activity (Figure 10).
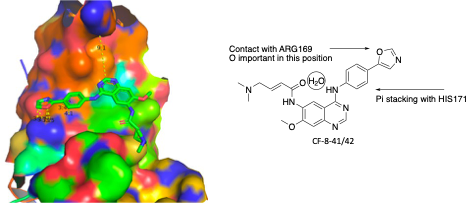
Figure 9. Interaction of covalent inhibitor CF-8-41 at the TBXT pocket.
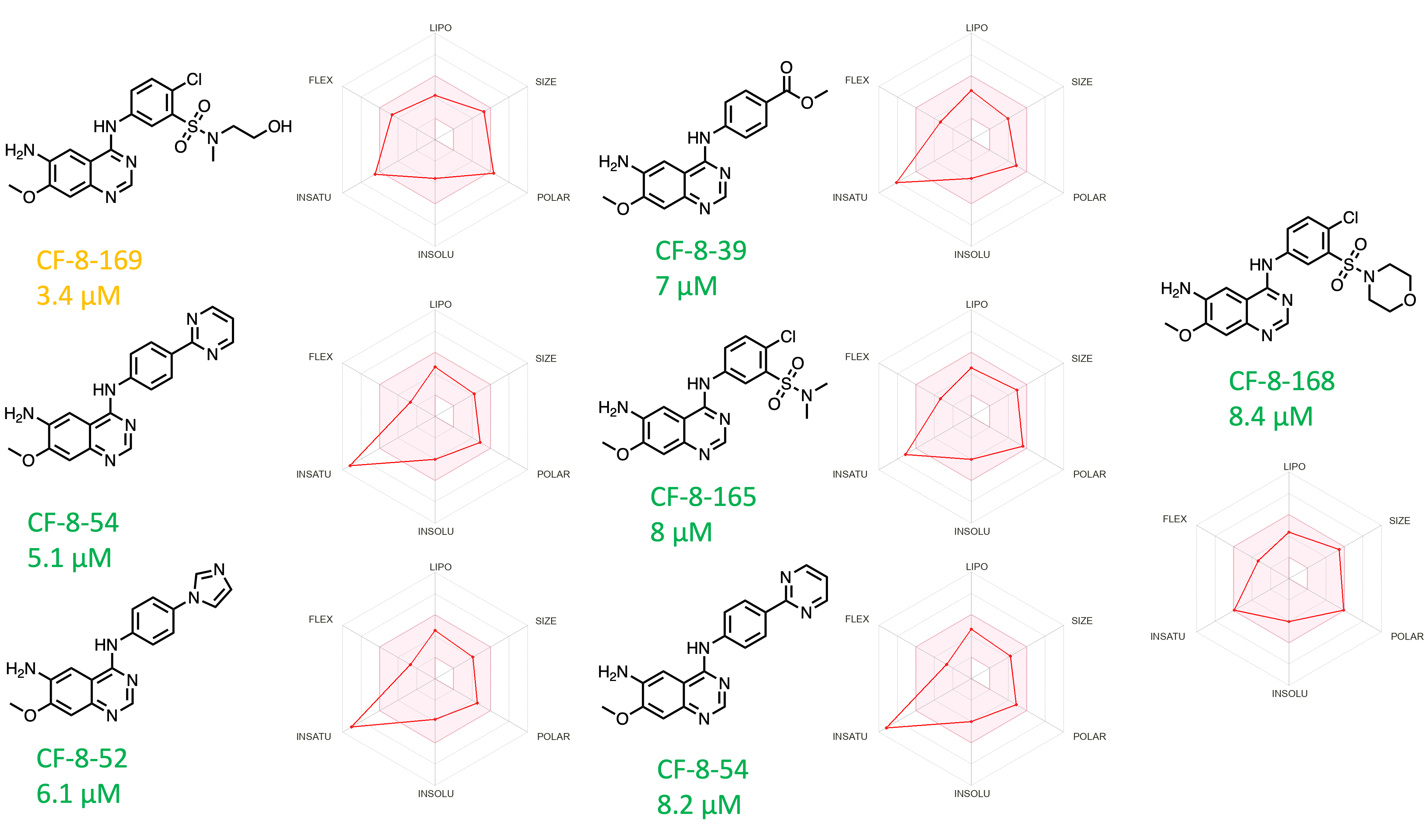
Figure 10. Quinazoline series with respective SPR Kd. [Color code from manual data analysis and visualization: Green = Excellent Curve Fitting Data; Orange = Good Curve Fitting Data with outlier data point; Red = Not a good curve fitting data; Radar Chart represent predicted ADME properties using SWISSADME]
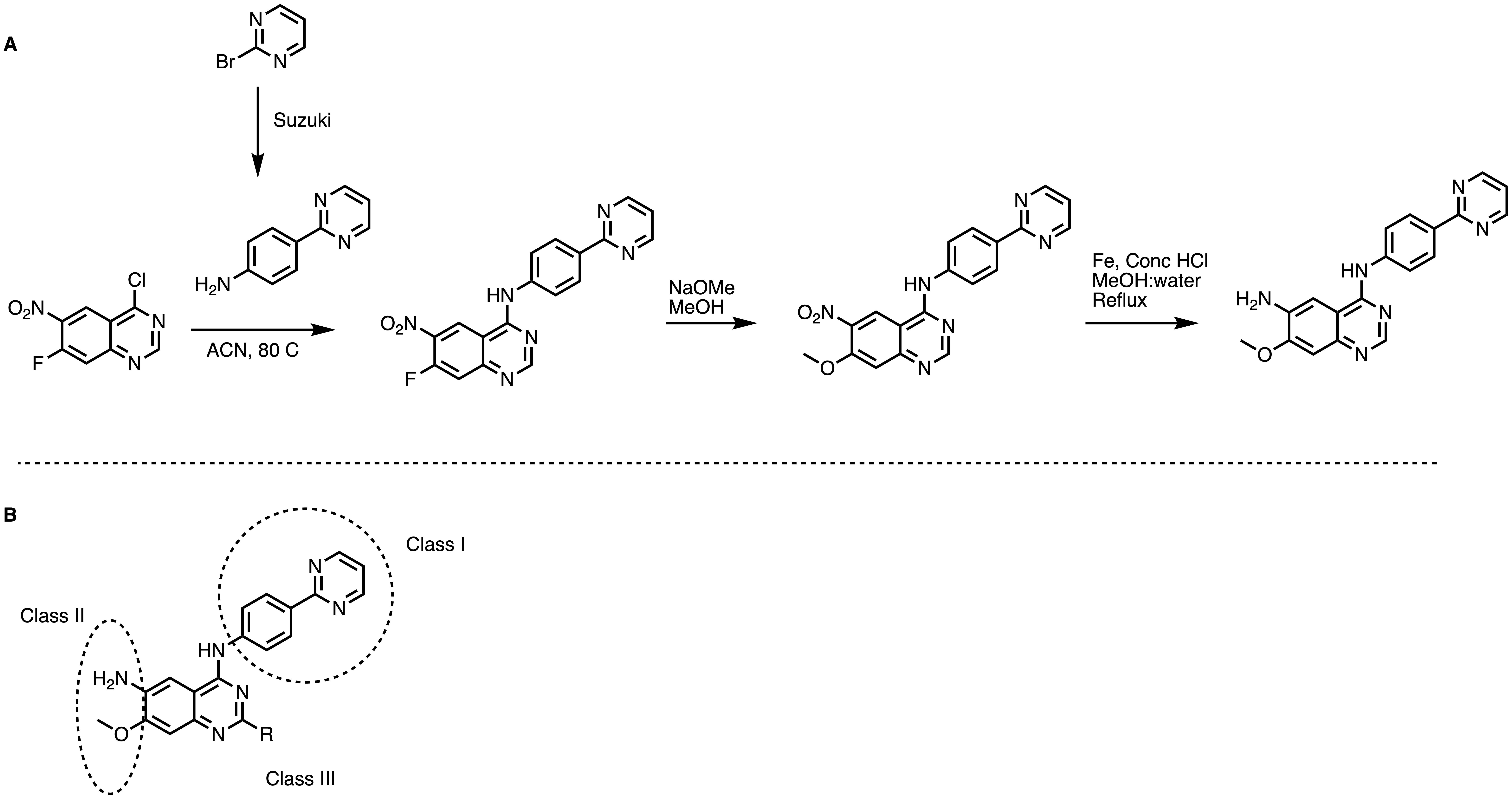
Figure 11. General synthetic scheme for quinazolines (A). SAR Strategies (B)
Key properties of the quinazoline series:
- SPR activity for multiple examples from the scaffold (7 of 34 analogs < 10 μM)
- Moderate molecular weight (Avg. <350)
- efficient synthesis (Figure 11A)
- Potential for covalent inhibitors and bivalent degrader development
Challenges to overcome: solubility, kinase activity
In summary:

Representatives from the top 3 prioritized series